What You Will Learn in This Article
- Trace the complete path of dietary iron from the intestinal lumen to haemoglobin synthesis
- Name every key protein in iron absorption, transport, and storage with its function
- Explain the hepcidin-ferroportin regulatory axis — the master controller of systemic iron homeostasis
- Map total body iron distribution and daily iron balance
- Connect iron to haem synthesis — all 8 enzymatic steps, locations, and enzyme defects
- Distinguish iron deficiency anaemia from anaemia of chronic disease using laboratory parameters
- Identify hereditary haemochromatosis, sideroblastic anaemia, and lead poisoning by their biochemical signatures
- Apply laboratory investigations to diagnose iron disorders correctly
- Explain oral iron pharmacology, parenteral iron, and chelation therapy
📖 Introduction: Why This Topic Matters in Exams
A 28-year-old woman presents with fatigue, pallor, pica (craving for ice — pagophagia), and brittle spoon-shaped nails. Her haemoglobin is 7.2 g/dL with an MCV of 62 fL. Serum iron is low, total iron-binding capacity (TIBC) is elevated, and serum ferritin is 4 ng/mL. She has iron deficiency anaemia — the most common nutritional deficiency worldwide and the most common cause of anaemia in clinical practice. Every laboratory value makes mechanistic sense: low ferritin = depleted stores; low serum iron = insufficient supply; high TIBC = upregulated transferrin synthesis to compensate; microcytic hypochromic anaemia = insufficient haem for haemoglobin.
Iron metabolism appears in medical entrance exams across every discipline: biochemistry (absorption machinery, haem synthesis), physiology (hepcidin regulation, iron balance), haematology (iron deficiency anaemia, anaemia of chronic disease, haemochromatosis), pharmacology (oral iron, chelation therapy), and pathology (porphyrias, sideroblastic anaemia). Understanding iron as a unified story — from duodenal crypt cell to bone marrow erythroblast — allows you to reason through any clinical scenario.
🔬 Section 1 — Iron Chemistry and Body Distribution
1.1 Why Iron Is Essential — and Dangerous
Iron cycles between two oxidation states:
- Fe²⁺ (ferrous): Reduced, soluble, transportable, absorbed
- Fe³⁺ (ferric): Oxidised, insoluble at physiological pH, stored, protein-bound
This redox cycling is essential for:
- Oxygen transport (haemoglobin Fe²⁺ binds O₂)
- Electron transport chain (iron-sulfur clusters, haem cytochromes)
- Enzymatic catalysis (aconitase, ribonucleotide reductase)
- DNA synthesis (ribonucleotide reductase requires iron)
The same redox chemistry creates danger via the Fenton reaction:
Fe²⁺ + H₂O₂ → Fe³⁺ + OH• + OH⁻
The hydroxyl radical (OH•) generated is one of the most reactive oxidants known — damaging lipids (lipid peroxidation), proteins, and DNA. This is why iron is NEVER found free in the body — it is always protein-bound.

Figure 2: The Fenton reaction — why free iron is toxic. Fe²⁺ catalyses hydroxyl radical production from H₂O₂, causing oxidative damage. Iron-binding proteins (transferrin, ferritin) prevent free iron from participating in this reaction.
1.2 Total Body Iron Distribution
Total body iron: ~3.5–4 g (males); ~2.5–3 g (females)
| Compartment | Amount | % of Total | Form |
|---|---|---|---|
| Haemoglobin (in RBCs) | ~2,500 mg | ~65% | Haem-Fe²⁺ |
| Storage (liver, spleen, bone marrow) | ~1,000 mg | ~25% | Ferritin + Haemosiderin |
| Myoglobin (muscle) | ~300 mg | ~8% | Haem-Fe²⁺ |
| Tissue enzymes (cytochromes, etc.) | ~150 mg | ~4% | Various |
| Plasma (transferrin-bound) | ~3–4 mg | ~0.1% | Fe³⁺ on transferrin |
Key insight: Despite being only 0.1% of body iron, the transferrin pool turns over ~10 times per day — it is the most dynamic iron compartment. The ~3 mg of transferrin-bound iron is replenished by macrophage recycling of senescent RBCs (~20–25 mg/day) and dietary absorption (~1–2 mg/day).
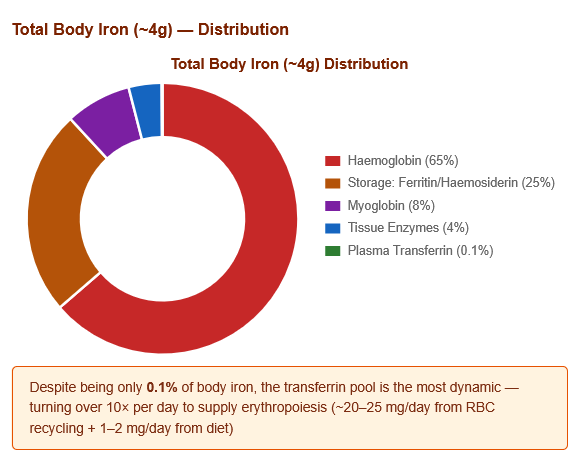
Figure 3: Total body iron distribution. Haemoglobin dominates (65%). Despite being a tiny fraction (0.1%), plasma transferrin is the most dynamic compartment — turning over 10× per day.
1.3 Daily Iron Balance
- Daily requirement absorbed: 1–2 mg
- Dietary intake: 10–20 mg/day (only ~10% absorbed)
- Daily losses: ~1–2 mg (desquamation, sweat, GI losses)
- NO active excretion mechanism — iron regulation occurs entirely at the level of absorption
- Increased requirements: Pregnancy (27 mg/day dietary need), menstruating women (18 mg/day), infants/adolescents (growth)
🔬 Section 2 — Iron Absorption: Step-by-Step Molecular Machinery
2.1 Forms of Dietary Iron
| Feature | Haem Iron | Non-Haem Iron |
|---|---|---|
| Sources | Meat, poultry, fish (myoglobin + Hb) | Plants, dairy, eggs, fortified foods |
| Chemical form | Fe²⁺ in porphyrin ring | Fe³⁺ (insoluble) |
| Absorption | 20–30% | 2–10% |
| Transporter | HCP1 (Haem Carrier Protein 1) | Requires DcytB reduction then DMT1 |
| Effect of diet | Not affected by inhibitors | Inhibited by phytates, oxalates, tannins, calcium |
| Enhancers | — | Vitamin C, citric acid, meat factor |
2.2 Absorption Site
Iron is absorbed exclusively in the duodenum and proximal jejunum — the first 20–30 cm of small intestine.
- Lowest intestinal pH (gastric acid) → iron stays soluble
- Highest density of absorption proteins (DcytB, DMT1, ferroportin)
- Duodenal crypt cells sense body iron stores → programme their iron-absorbing capacity before migrating up the villus
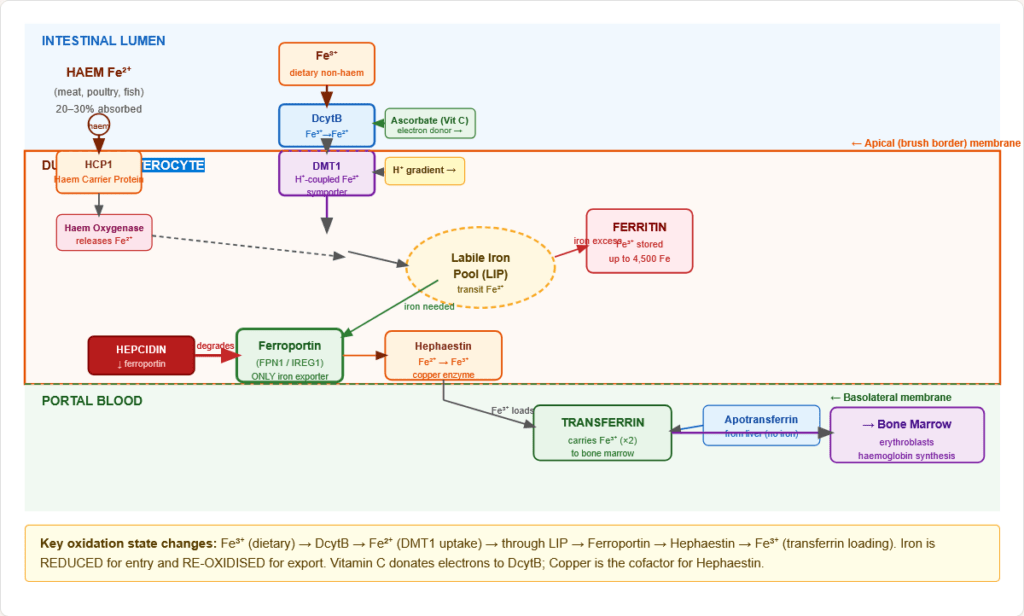
Figure 4: Anatomical localisation of iron absorption. The duodenum and proximal jejunum are the exclusive sites. The crypt-to-villus migration of enterocytes allows dynamic programming of absorption capacity in response to body iron status.
2.3 Complete Non-Haem Iron Absorption — Molecular Steps
Step 1 — Reduction at the brush border:
- DcytB (Duodenal Cytochrome B): Brush-border ferrireductase on the apical membrane
- Reduces Fe³⁺ → Fe²⁺ using ascorbate (Vitamin C) as electron donor
- Molecular explanation for why Vitamin C enhances iron absorption — it donates electrons to DcytB
- Also regulated by hypoxia (HIF-2α upregulates DcytB and DMT1)
Step 2 — Apical uptake via DMT1:
- DMT1 (Divalent Metal Transporter 1 / SLC11A2 / Nramp2):
- Proton-coupled symporter (H⁺:Fe²⁺ = 1:1)
- Transports Fe²⁺ into the enterocyte — and other divalent metals (Mn²⁺, Zn²⁺, Cu²⁺, Pb²⁺, Cd²⁺)
- Located on apical (luminal) membrane
- Upregulated by iron deficiency, hypoxia (HIF-2α), and low pH
- The H⁺ gradient explains why low gastric pH (acidic duodenum) enhances iron absorption; PPI/antacid use impairs absorption
Step 3 — Intracellular transit:
- Fe²⁺ enters the labile iron pool (LIP) — a transit pool of chelated iron
- From the LIP, iron can be: stored as ferritin (if replete) OR exported basolaterally (if needed)
Step 4 — Basolateral export via Ferroportin:
- Ferroportin (FPN1 / SLC40A1 / IREG1):
- The ONLY known iron exporter in mammalian cells — there is no alternative
- On the basolateral membrane of enterocytes, macrophages, hepatocytes, placental trophoblasts
- Transports Fe²⁺ out of the cell into portal blood
- Inhibited by hepcidin — the master regulatory hormone (hepcidin binds ferroportin → internalisation → lysosomal degradation)
Step 5 — Oxidation and loading onto transferrin:
- Hephaestin (HEPH):
- Copper-dependent ferroxidase at the basolateral membrane
- Oxidises Fe²⁺ → Fe³⁺ (only Fe³⁺ binds transferrin)
- Structural analogue of ceruloplasmin (the plasma ferroxidase)
- Copper deficiency → ↓ hephaestin → functional iron deficiency even with adequate iron
- Fe³⁺ loads onto apotransferrin in portal blood → transferrin-Fe complex circulates to tissues
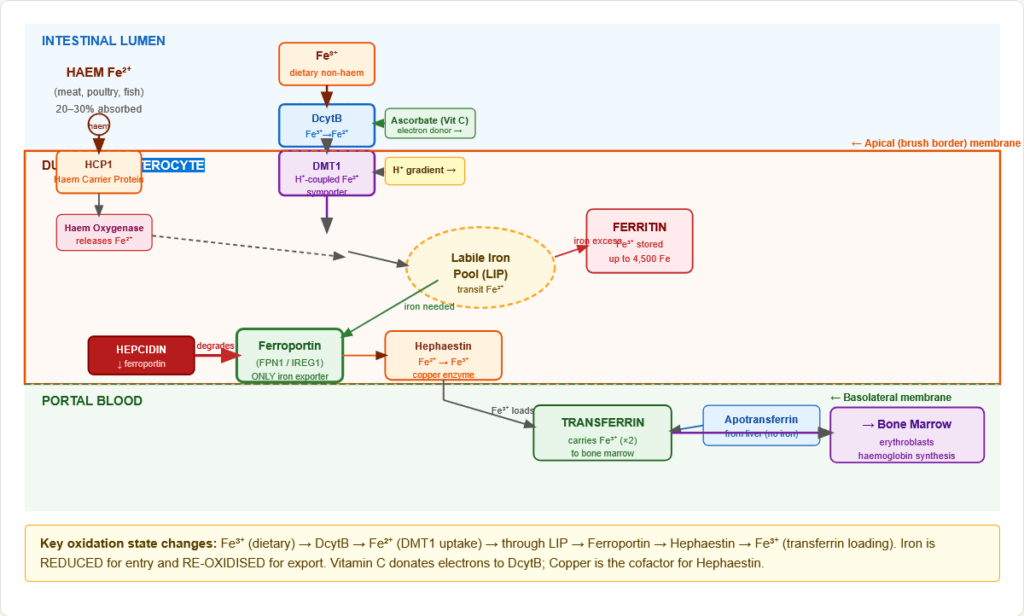
Figure 5: Complete molecular machinery of iron absorption in the duodenal enterocyte. Key: iron changes oxidation state twice — reduced apically by DcytB (Fe³⁺→Fe²⁺) and re-oxidised basally by hephaestin (Fe²⁺→Fe³⁺) for transferrin loading. Hepcidin controls the ferroportin gate.
⚙️ Section 3 — Iron Transport: The Transferrin Receptor Cycle
3.1 Transferrin — The Iron Taxi
Transferrin:
- Glycoprotein (~80 kDa) synthesised primarily in the liver
- Contains two iron-binding sites (N-lobe + C-lobe) — binds Fe³⁺ with very high affinity (Ka ~10²³ M⁻¹)
- Keeps iron soluble, non-toxic, and readily deliverable
- Transferrin saturation (%) = (serum iron ÷ TIBC) × 100 — normal 20–45%
- Forms: Apotransferrin (0 Fe), Monoferric (1 Fe), Diferric (2 Fe — binds TfR1 most avidly)
3.2 The Transferrin Receptor Cycle (TfR1 Cycle)
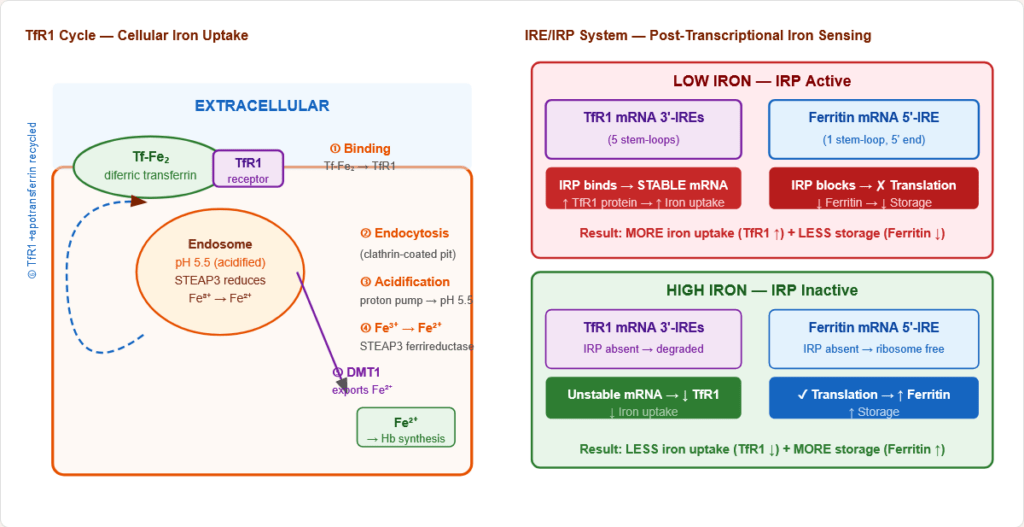
Figure 6: The transferrin receptor (TfR1) cycle — the universal mechanism of cellular iron uptake. Diferric transferrin is endocytosed; iron is released at acidic pH; apotransferrin-TfR1 is recycled to the cell surface. This cycle repeats ~10–20 times per minute in active erythroblasts.
Step-by-step:
- Diferric transferrin binds TfR1 on cell surface
- Complex internalised by clathrin-mediated endocytosis
- Endosome acidified to pH ~5.5 by proton pump → Fe³⁺ released from transferrin
- STEAP3 (endosomal ferrireductase) reduces Fe³⁺ → Fe²⁺
- DMT1 transports Fe²⁺ out of endosome into cytoplasm
- Apotransferrin + TfR1 recycled to cell surface; apotransferrin dissociates at neutral pH
3.3 The IRE/IRP System — Post-Transcriptional Regulation
This elegant system controls TfR1 and ferritin expression reciprocally based on intracellular iron levels.
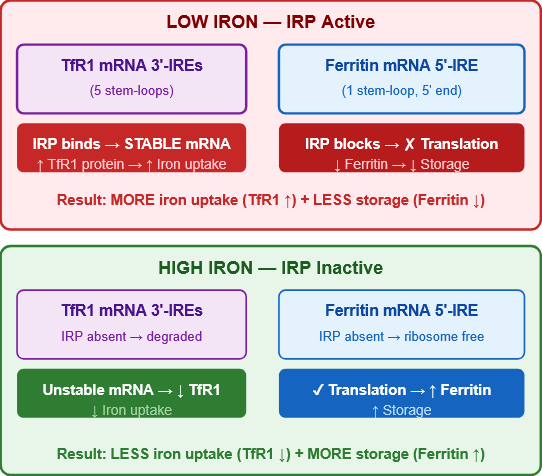
Figure 7: The IRE/IRP post-transcriptional regulatory system. TfR1 mRNA has five IRE stem-loops in its 3′-UTR; ferritin mRNA has one IRE in its 5′-UTR. IRP activity (high in iron deficiency) stabilises TfR1 mRNA and blocks ferritin translation — maximising iron uptake and minimising storage. Elegant reciprocal control.
| Condition | IRP activity | TfR1 mRNA | TfR1 protein | Ferritin mRNA | Ferritin protein |
|---|---|---|---|---|---|
| Low iron | High (binds IREs) | Stabilised | ↑ (more iron uptake) | Translation blocked | ↓ (less storage) |
| High iron | Low (IRP inactive) | Destabilised | ↓ (less uptake) | Translated freely | ↑ (more storage) |
🔬 Section 4 — Iron Storage: Ferritin and Haemosiderin
4.1 Ferritin — Primary Storage
Ferritin structure:
- Hollow spherical protein — 24 subunits of two types:
- H-chain (Heavy): Has ferroxidase activity — oxidises Fe²⁺→Fe³⁺ for storage; predominant in heart
- L-chain (Light): Structural stability; nucleation of iron core; predominant in liver/spleen
- Stores up to 4,500 iron atoms as Fe(O)OH (ferric oxyhydroxide) in the hollow interior
- Serum ferritin: Reflects body iron stores (1 ng/mL ≈ 10 mg stored iron)
- Normal: 12–300 ng/mL (men); 12–150 ng/mL (women)
- <12 ng/mL = iron deficiency (most specific single test for iron depletion)
- Ferritin is an acute-phase reactant — can be falsely normal/elevated in inflammation, infection, liver disease, malignancy even with true iron deficiency
Figure 8: Ferritin molecular architecture. The 24-subunit hollow apoferritin shell stores up to 4,500 Fe³⁺ ions. H-chains provide ferroxidase activity (Fe²⁺→Fe³⁺ for storage); L-chains provide structural integrity. Haemosiderin (right) is insoluble aggregated ferritin formed under iron overload.
4.2 Haemosiderin — Secondary Storage
- Partially degraded ferritin shells with aggregated iron — visible as golden-brown granules (Prussian blue stain)
- Less readily mobilisable than ferritin
- Seen in iron overload (haemochromatosis, transfusional iron overload)
- Bone marrow Prussian blue staining assesses iron stores in macrophages
⚙️ Section 5 — Hepcidin: The Master Regulator
5.1 What Is Hepcidin?
Hepcidin is a 25-amino acid peptide hormone synthesised by hepatocytes. It is the master regulator of systemic iron homeostasis.
Mechanism of action:
- Hepcidin binds ferroportin on enterocytes, macrophages, and hepatocytes
- Ferroportin-hepcidin complex is ubiquitinated → internalised → lysosomal degradation
- Without ferroportin: iron cannot exit cells → trapped in enterocytes (↓ absorption), macrophages (↓ iron recycling), hepatocytes (↓ store release)
- Net result: ↑ hepcidin = ↓ serum iron (iron sequestration)
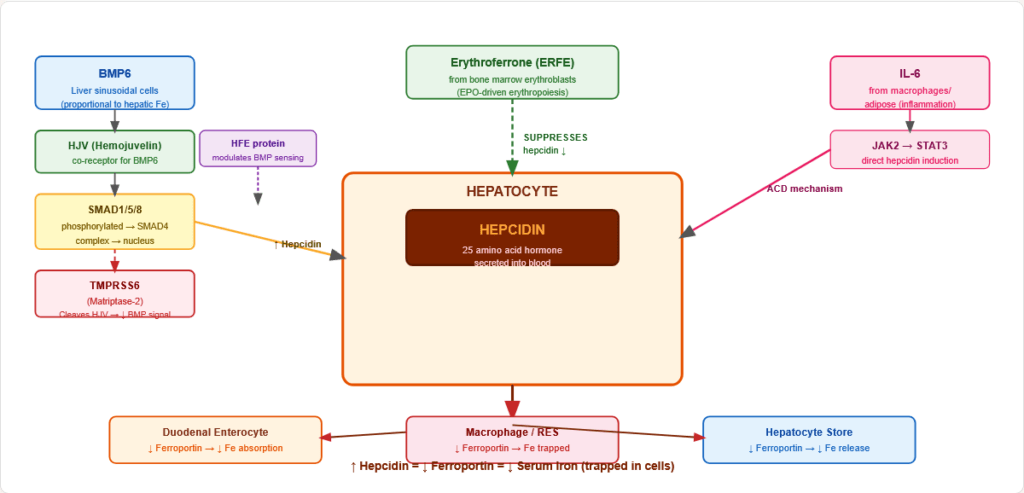
Figure 9: The hepcidin-ferroportin axis — the master iron regulatory switch. High hepcidin (infection, iron overload) traps iron in cells. Low hepcidin (iron deficiency, hypoxia, anaemia) allows maximal iron export via ferroportin.
5.2 Regulation of Hepcidin Expression
Hepcidin is INCREASED by:
- Iron overload (via BMP6-SMAD signalling from liver sinusoidal cells)
- Inflammation/infection (IL-6 → JAK2 → STAT3 → hepcidin gene)
- Obesity (elevated IL-6)
Hepcidin is DECREASED by:
- Iron deficiency
- Hypoxia (HIF-2α → TMPRSS6 → cleaves HJV → ↓ BMP signalling)
- Anaemia / increased erythropoiesis (erythroferrone — ERFE — from erythroblasts suppresses hepcidin)
- Haemochromatosis mutations (HFE, HJV, HAMP defects → hepcidin deficiency)
The BMP6/SMAD molecular pathway:
- BMP6 (from liver sinusoidal cells, proportional to hepatic iron) → binds BMPR + HJV co-receptor → phosphorylates SMAD1/5/8 → SMAD4 complex → hepcidin gene transcription
- TMPRSS6 (matriptase-2): Cleaves HJV → reduces BMP signalling → reduces hepcidin → used in iron refractory iron deficiency anaemia (IRIDA — loss of TMPRSS6 → ↑ hepcidin → refractory IDA)
- HFE mutation (C282Y) → impaired BMP sensing → ↓ hepcidin → haemochromatosis
Figure 10: Molecular map of hepcidin regulation. Multiple signals converge on the hepcidin promoter: BMP6/SMAD (iron sensor), IL-6/STAT3 (inflammation), TMPRSS6/HJV (negative regulator), and erythroferrone (erythropoiesis signal). The integration of these inputs determines systemic iron availability.
🔬 Section 6 — Haem Synthesis: 8 Enzymatic Steps
6.1 Overview
Haem = iron-protoporphyrin IX. Synthesised in all nucleated cells; highest in:
- Bone marrow erythroblasts (~85%) — for haemoglobin
- Liver hepatocytes — for cytochrome P450 and other haem-proteins
Overall equation: 8 Succinyl-CoA + 8 Glycine → Protoporphyrin IX → + Fe²⁺ → Haem
6.2 The 8 Steps in Detail
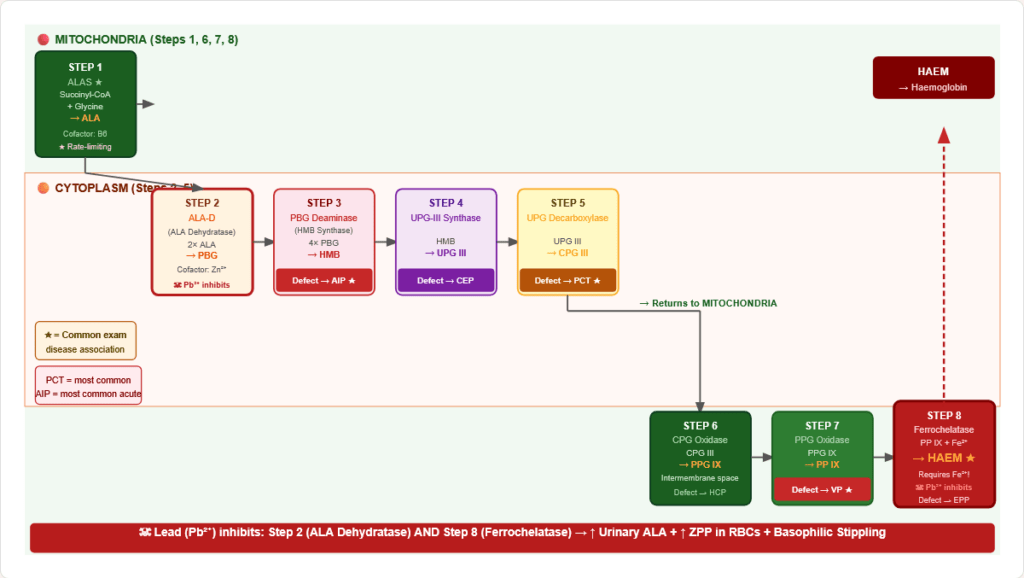
Figure 11: Complete haem synthesis pathway with subcellular localisation. ALAS (step 1) is rate-limiting. Lead inhibits steps 2 (ALA-D) and 8 (ferrochelatase). Clinical defects at each step cause specific porphyrias.
| Step | Substrate → Product | Enzyme | Location | Cofactor | Deficiency / Inhibitor |
|---|---|---|---|---|---|
| 1 | Succinyl-CoA + Glycine → ALA | ALAS1/ALAS2 ★ rate-limiting | Mitochondria | B6 (PLP) | ALAS2 mutations → X-linked sideroblastic anaemia |
| 2 | 2 ALA → Porphobilinogen (PBG) | ALA Dehydratase (ALA-D) | Cytoplasm | Zn²⁺ | Lead (Pb²⁺) displaces Zn²⁺ → inhibited |
| 3 | 4 PBG → Hydroxymethylbilane | PBG Deaminase (HMB synthase) | Cytoplasm | — | Deficiency → Acute Intermittent Porphyria (AIP) |
| 4 | HMB → Uroporphyrinogen III | UPG-III Synthase | Cytoplasm | — | Deficiency → Congenital Erythropoietic Porphyria (CEP) |
| 5 | UPG-III → Coproporphyrinogen III | Uroporphyrinogen Decarboxylase | Cytoplasm | — | Deficiency → Porphyria Cutanea Tarda (PCT) — most common porphyria |
| 6 | CPG-III → Protoporphyrinogen IX | Coproporphyrinogen Oxidase | Mito (inter-membrane) | O₂ | Deficiency → Hereditary Coproporphyria |
| 7 | PPG-IX → Protoporphyrin IX | Protoporphyrinogen Oxidase | Mitochondria | — | Deficiency → Variegate Porphyria (VP) |
| 8 | Protoporphyrin + Fe²⁺ → Haem | Ferrochelatase | Mitochondria | Fe²⁺ | Lead (Pb²⁺) inhibits; Deficiency → EPP; requires Fe²⁺ not Fe³⁺ |
6.3 ALAS Regulation — The Two Isoforms
ALAS1 (liver, housekeeping):
- Feedback-inhibited by haem (product inhibition)
- Induced by: drugs (barbiturates, rifampicin, alcohol, oestrogens) — all increase CYP450 demand → consume haem → ↓ haem pool → ↑ ALAS1 → toxic porphyrin intermediates if downstream enzyme partially deficient
- Drug-induced porphyria attacks explained by ALAS1 induction
ALAS2 (erythroid-specific):
- Has 5′-IRE in mRNA → regulated by IRP/iron status
- Iron deficiency → IRP blocks ALAS2 translation → ↓ haem synthesis in erythroblasts
- ALAS2 mutations → X-linked sideroblastic anaemia (XSA) — pyridoxine-responsive if residual enzyme activity
🏥 Section 7 — Clinical Disorders of Iron Metabolism
7.1 Iron Deficiency Anaemia (IDA)
Most common nutritional deficiency and most common cause of anaemia worldwide
Progressive stages:
| Stage | Iron Stores | Serum Iron | TIBC | Tf Saturation | Ferritin | Hb | Blood Film |
|---|---|---|---|---|---|---|---|
| 1 — Iron depletion | ↓↓ | Normal | Normal | Normal | ↓ (<12) | Normal | Normal |
| 2 — Iron-deficient erythropoiesis | Absent | ↓ | ↑ | ↓ | ↓↓ | Normal | Normal |
| 3 — Iron deficiency anaemia | Absent | ↓↓ | ↑↑ | ↓↓ (<15%) | ↓↓↓ | ↓ | Microcytic hypochromic |
Causes:
- Increased demand: pregnancy, rapid growth (infancy, adolescence)
- Decreased intake: vegetarian/vegan diet, malnutrition
- Malabsorption: coeliac disease (duodenal villi damaged), H. pylori (achlorhydria), post-gastrectomy, PPIs
- Chronic blood loss: menorrhagia (#1 in premenopausal women), GI bleeding (peptic ulcer, CRC, hookworm), haematuria
Specific clinical signs of IDA:
- Koilonychia (spoon-shaped nails)
- Angular cheilitis (mouth corner fissures)
- Glossitis (smooth, painful tongue)
- Pica (pagophagia = ice; geophagia = soil; amylophagia = starch)
- Plummer-Vinson / Paterson-Brown-Kelly syndrome = IDA + oesophageal web + dysphagia (pre-malignant — risk of post-cricoid carcinoma)
7.2 Anaemia of Chronic Disease (ACD)
Second most common cause of anaemia; caused by hepcidin-mediated iron sequestration
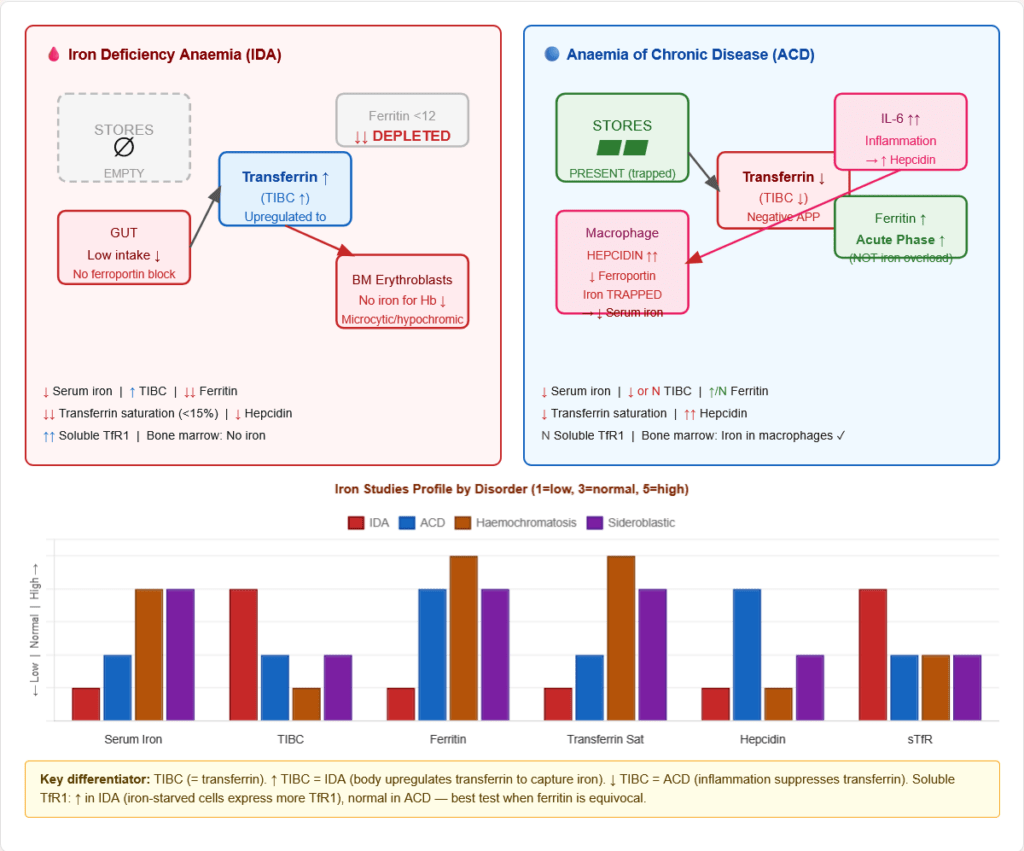
Figure 12: Iron flow in IDA vs ACD. Key distinction: in IDA, stores are depleted (↓ ferritin, ↑ TIBC). In ACD, iron is present but trapped by elevated hepcidin (↑ ferritin, ↓ TIBC, iron visible in BM macrophages but not in erythroblasts).
Mechanism:
- Chronic disease (infection, cancer, autoimmune, CKD, RA)
- Inflammatory cytokines (IL-6, IL-1β, TNF-α) released
- IL-6 → STAT3 → ↑ hepcidin production by liver
- ↑ Hepcidin → ↓ ferroportin on macrophages → iron trapped → ↓ serum iron
- IL-1β → ↓ EPO production (kidney) + blunted EPO response in bone marrow
- Result: Normocytic normochromic anaemia with elevated ferritin
Laboratory comparison:
| Parameter | IDA | ACD |
|---|---|---|
| Serum iron | ↓↓ | ↓ |
| TIBC | ↑↑ | ↓ or Normal |
| Transferrin saturation | ↓↓ (<15%) | ↓ (15–20%) |
| Serum ferritin | ↓↓ | ↑ or Normal |
| Serum hepcidin | ↓ | ↑↑ |
| Soluble TfR (sTfR) | ↑↑ | Normal |
| Bone marrow iron | Absent | Present (macrophages) |
7.3 Hereditary Haemochromatosis (HH)
Most common inherited metabolic disease in Northern Europeans (prevalence ~1:200 in Celtic populations)
Types:
| Type | Gene | Inheritance | Key Feature |
|---|---|---|---|
| Type 1 (HFE) | HFE — C282Y mutation (~85–90%) | AR | Most common; adult onset; liver + joints + diabetes |
| Type 2A (Juvenile) | HJV (Hemojuvelin) | AR | Severe; cardiomyopathy in teens/20s |
| Type 2B (Juvenile) | HAMP (Hepcidin) | AR | Most severe; juvenile onset |
| Type 3 | TFR2 | AR | Intermediate |
| Type 4A | SLC40A1 (ferroportin LOF) | AD | Macrophage-predominant loading |
| Type 4B | SLC40A1 (ferroportin GOF) | AD | Hepcidin-resistant; similar to type 1 |
HFE Pathophysiology: C282Y disrupts disulphide bond → misfolded HFE → impaired BMP6/SMAD sensing → ↓ hepcidin → ↑ ferroportin → excessive iron absorption (~3–4 mg/day vs normal 1–2 mg) → iron accumulation over decades
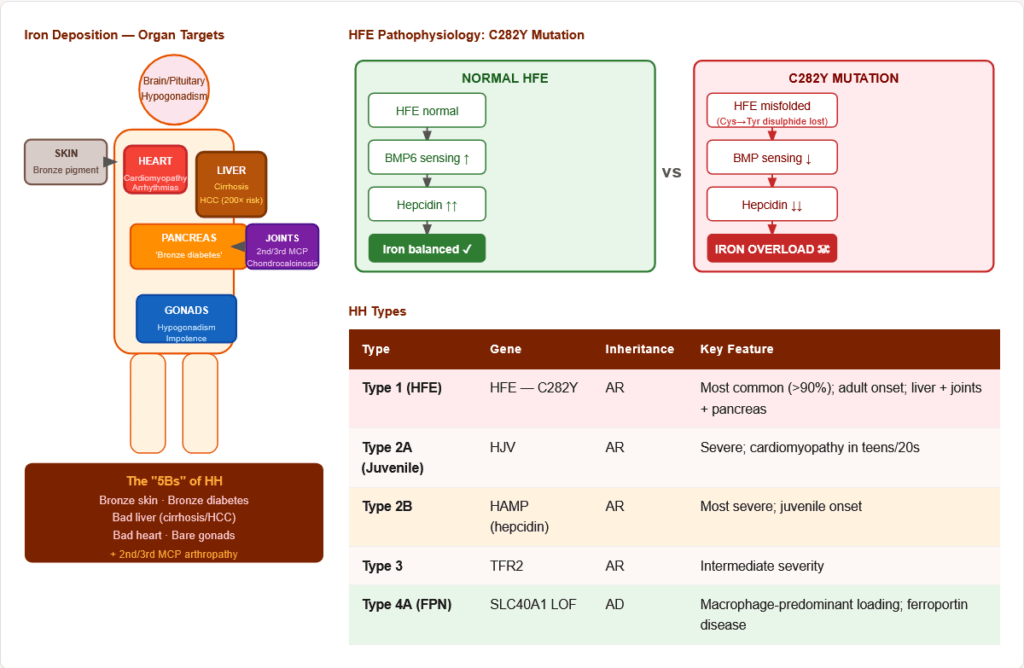
Figure 13: Organ involvement in hereditary haemochromatosis — the “Bronze Diabetic.” Iron deposits accumulate over decades in liver (→ cirrhosis, HCC — 200× risk), pancreas (→ insulin-dependent diabetes), heart (→ dilated cardiomyopathy), skin (→ bronze pigmentation), MCP joints (→ chondrocalcinosis), and pituitary/gonads (→ hypogonadism).
Clinical features — “5Bs”: Bronze skin · Bronze diabetes · Bad liver (cirrhosis/HCC) · Bad heart (cardiomyopathy) · Bare gonads (hypogonadism) + Joint arthropathy (2nd/3rd MCP joints — characteristic)
Investigations:
- Fasting transferrin saturation >45% — best initial screening test
- Ferritin >300 ng/mL (men) / >200 ng/mL (women)
- HFE gene testing: C282Y homozygous, or C282Y/H63D compound heterozygous
- MRI liver (T2* imaging — non-invasive iron quantification)
- Liver biopsy: Perls’ Prussian blue stain; hepatic iron index >1.9
Treatment:
- Phlebotomy (first-line): Weekly 450–500 mL blood removal (250 mg iron/unit) until ferritin <50 ng/mL; then maintenance every 2–4 months
- Avoid: alcohol (hepatotoxic), Vitamin C supplements (mobilises iron → oxidative burst), raw shellfish (Vibrio vulnificus — thrives in iron-rich environments)
7.4 Sideroblastic Anaemia
Characterised by ring sideroblasts in bone marrow — erythroblasts with iron-laden mitochondria forming a ring around the nucleus (Prussian blue stain).
Pathophysiology: Defect in haem synthesis → iron accumulates in mitochondria (cannot be incorporated into protoporphyrin) → ring sideroblasts
Causes:
- Congenital: ALAS2 mutations (X-linked sideroblastic anaemia), SLC25A38 mutations, Pearson syndrome (mitochondrial DNA deletion)
- Acquired:
- Lead poisoning (inhibits ALA-D + Ferrochelatase → ring sideroblasts + basophilic stippling)
- Isoniazid / anti-TB drugs (B6 antagonism → ↓ ALAS2 cofactor)
- Alcohol (mitochondrial toxicity + B6 antagonism)
- Chloramphenicol (mitochondrial RNA polymerase inhibition)
- MDS with ring sideroblasts (SF3B1 mutation — most common adult form)
Labs: ↑ Serum iron; ↑ Ferritin; Normal/↓ TIBC; Dimorphic blood film; Ring sideroblasts >15% on BM aspirate (Prussian blue)
7.5 Lead Poisoning — Haem Synthesis Targets
Lead has two specific targets in haem synthesis:
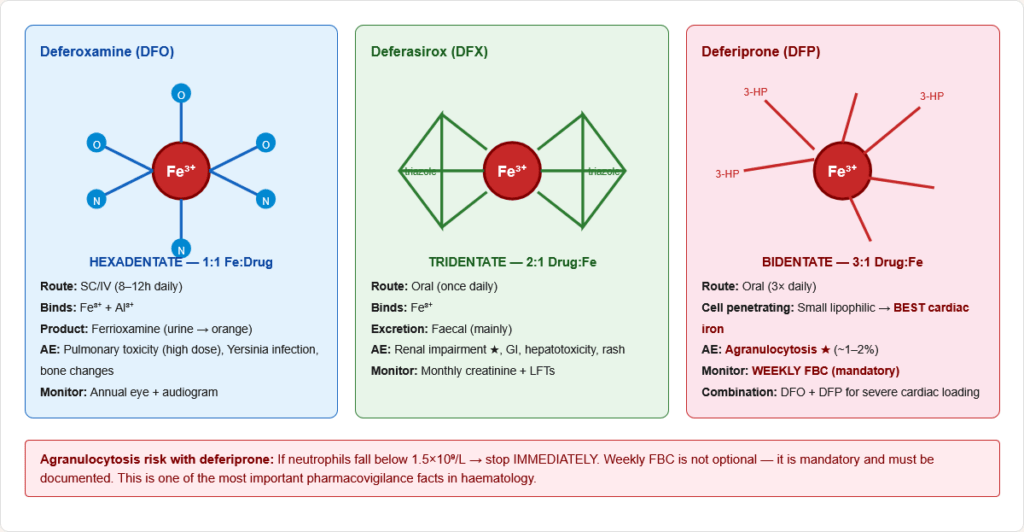
Figure 14: Two specific lead targets in haem synthesis. (1) ALA Dehydratase (step 2) — Pb²⁺ displaces Zn²⁺ → ALA accumulates → ↑ urinary ALA. (2) Ferrochelatase (step 8) — Pb²⁺ inhibits Fe²⁺ insertion → zinc substitutes → ZPP (zinc protoporphyrin) accumulates in RBCs. Both markers are diagnostically valuable.
- ALA Dehydratase (Step 2): Pb²⁺ displaces Zn²⁺ from active site → inhibits ALA→PBG → ↑ urinary ALA
- Ferrochelatase (Step 8): Pb²⁺ inhibits → zinc substitutes for iron → ↑ ZPP (zinc protoporphyrin) in RBCs
Blood film: Basophilic stippling — aggregated ribosomes visible as blue dots in RBCs (pathognomonic for lead poisoning)
🔬 Section 8 — Laboratory Interpretation
8.1 The Complete Iron Panel Interpretation
Figure 15: Clinical algorithm for iron studies interpretation. Start with serum ferritin (most specific for iron stores); proceed to TIBC and transferrin saturation; use sTfR to distinguish IDA from ACD when ferritin is equivocal. Remember ferritin is an acute-phase reactant.
Decision framework:
| Ferritin | TIBC | Tf Sat | Likely diagnosis |
|---|---|---|---|
| ↓↓ (<12) | ↑ | ↓↓ | Iron Deficiency Anaemia |
| Normal/↑ | ↓ | ↓ | ACD (hepcidin-mediated) |
| ↓ + elevated CRP | ↓ or normal | ↓ | ACD + IDA (concurrent) → use sTfR |
| ↑↑ | ↓ | ↑↑ (>50%) | Haemochromatosis or iron overload |
| ↑ | ↓ or N | ↑ | Sideroblastic anaemia |
8.2 Zinc Protoporphyrin (ZPP) / Free Erythrocyte Protoporphyrin (FEP)
- When Fe²⁺ is unavailable, ferrochelatase inserts zinc instead → ZPP
- Elevated in: IDA, lead poisoning, chronic disease
- ZPP/haem ratio >80 µmol/mol haem → iron-deficient erythropoiesis
- Useful screening in children (portable instruments available)
8.3 Soluble Transferrin Receptor (sTfR)
- TfR1 is shed from cell surfaces → sTfR in serum reflects TfR1 expression
- Elevated in iron deficiency (iron-starved cells express more TfR1)
- Normal/low in ACD (adequate iron in cells — hepcidin trapping, not absent iron)
- Best single test to distinguish IDA from ACD in the presence of inflammation
- sTfR/log Ferritin index >2 = iron deficiency even with concurrent inflammation
🔬 Section 9 — Pharmacology of Iron
9.1 Oral Iron Therapy
Ferrous (Fe²⁺) salts preferred — better absorbed than ferric
| Preparation | Elemental Iron | Notes |
|---|---|---|
| Ferrous sulphate 325 mg | 65 mg | Most used; cheapest |
| Ferrous gluconate 325 mg | 36 mg | Gentler GI profile |
| Ferrous fumarate 325 mg | 106 mg | High elemental iron content |
Optimising absorption:
- Take on empty stomach (best absorption)
- Co-administer Vitamin C (enhances DcytB/ascorbate reduction)
- Separate by ≥2h from: PPIs, antacids, calcium, tetracyclines, fluoroquinolones, levothyroxine, bisphosphonates
Side effects: GI (nausea, constipation, dark stools, epigastric pain) — major cause of non-adherence
Monitoring response:
- Reticulocyte count peaks at 7–10 days — earliest sign of response
- Haemoglobin rises ~1 g/dL/week starting at 2–4 weeks
- Continue 3–6 months AFTER Hb normalises to replenish stores (target ferritin >50 ng/mL)
9.2 Parenteral Iron
Indications: Malabsorption, intolerance to oral iron, active bleeding, CKD on ESA therapy, IBD, pre-operative anaemia
| Preparation | Key Feature | Risk |
|---|---|---|
| Iron sucrose (Venofer) | Safest; multiple small doses | Minimal |
| Ferric carboxymaltose (Ferinject) | Single large dose up to 1000 mg | Low |
| Low MW iron dextran | Effective but older | Anaphylaxis (test dose needed) |
| Ferumoxytol | Rapid infusion; CKD | Hypotension |
9.3 Iron Chelation Therapy
Indications: Transfusional iron overload (thalassaemia major, sickle cell, MDS), HH when phlebotomy contraindicated
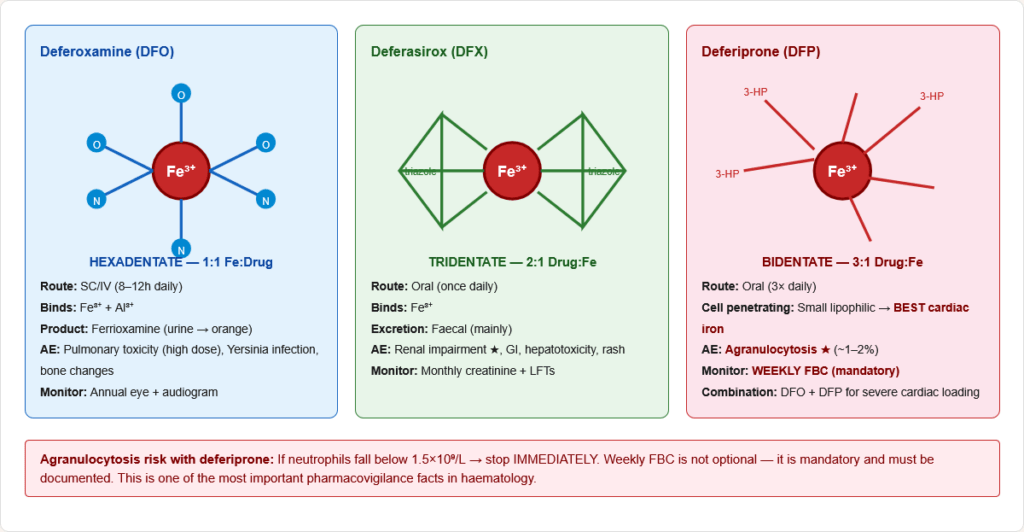
Figure 16: Chemical structures and iron-binding modes of the three clinical iron chelators. Deferoxamine (hexadentate, 1:1) binds Fe³⁺ with six coordination bonds; deferasirox (tridentate, 2:1) uses three bonds; deferiprone (bidentate, 3:1) uses two. Deferiprone’s small size enables cell membrane penetration — key for cardiac iron clearance.
| Drug | Route | Mechanism | Adverse Effect | Monitoring | Key Fact |
|---|---|---|---|---|---|
| Deferoxamine | SC/IV (8–12h infusion) | Hexadentate chelator; binds Fe³⁺ → ferrioxamine excreted in urine | Pulmonary toxicity (high dose), Yersinia infection, local reactions, bone changes | Annual eye + ear exam | Urine turns orange; original chelator; gold standard for severe overload |
| Deferasirox | Oral (once daily) | Tridentate; oral bioavailability; Fe³⁺ chelation | Renal impairment, GI upset, hepatotoxicity, rash | Monthly creatinine + LFTs | Most used oral chelator; revolutionised thalassaemia management |
| Deferiprone | Oral (3× daily) | Bidentate; crosses cell membranes | Agranulocytosis (~1–2%) — MUST monitor | Weekly FBC | Best cardiac iron clearance; combined with deferoxamine for severe cardiac iron loading |
🔄 Section 10 — Connections to Other Systems
→ Haemoglobin physiology — Iron (Fe²⁺ in haem) is the oxygen-binding atom. The proximal histidine coordinates haem iron; O₂ binds on the opposite face. Fe²⁺→Fe³⁺ oxidation = methaemoglobin → cannot carry O₂. Iron deficiency = ↓ haem = ↓ haemoglobin = microcytic hypochromic anaemia. Understanding iron directly explains haemoglobin function and pathology.
→ Porphyrias — All porphyrias are defects in haem synthesis enzymes. The clinical phenotype (acute neuropsychiatric attacks vs cutaneous photosensitivity) depends on which intermediates accumulate and where in the pathway. Acute porphyrias (AIP, VP, HCP) involve water-soluble intermediates that affect the nervous system; cutaneous porphyrias (PCT, CEP, EPP) involve photosensitising porphyrins.
→ Vitamin C metabolism — Vitamin C is the electron donor for DcytB (reduces Fe³⁺→Fe²⁺ for absorption) and prolyl/lysyl hydroxylases (collagen synthesis). Scurvy causes both impaired iron absorption and impaired collagen → perifollicular haemorrhages + poor wound healing.
→ Copper metabolism — Copper is required for both hephaestin (basolateral ferroxidase — exports iron from enterocytes) and ceruloplasmin (plasma ferroxidase). Copper deficiency → functional iron deficiency anaemia despite adequate iron stores — microcytic anaemia responding to copper supplementation, not iron.
→ Erythropoiesis and EPO — EPO (from renal interstitial fibroblasts) drives erythroid progenitor proliferation. Erythroblasts in response to EPO produce erythroferrone (ERFE), which suppresses hepcidin → mobilises iron for haemoglobin synthesis. In CKD: ↓ EPO + elevated hepcidin → combined deficiency. Iron must be repleted before or alongside ESA therapy.
→ Transferrin as negative acute-phase protein — Transferrin falls during inflammation (negative APP) while ferritin rises (positive APP). This creates the ACD laboratory pattern: ↓ TIBC, ↑ ferritin, ↓ serum iron. Misinterpretation of this pattern as iron overload is a critical diagnostic error to avoid.
🎯 High-Yield Exam Facts
🔴 Ferroportin is the ONLY mammalian iron exporter — hepcidin causes its degradation Hepcidin → ferroportin ubiquitination → lysosomal degradation → iron trapped. ↑ Hepcidin = ↓ serum iron. This single axis explains both ACD (IL-6 → ↑ hepcidin) and haemochromatosis (↓ hepcidin from HFE/HJV/HAMP mutations).
🔴 Serum ferritin <12 ng/mL = iron deficiency (virtually pathognomonic); but ferritin is an acute-phase reactant Ferritin can be falsely normal/elevated in iron deficiency concurrent with infection, inflammation, or liver disease. Use sTfR or bone marrow Prussian blue in this setting.
🔴 ALAS = rate-limiting enzyme of haem synthesis; ALAS2 (erythroid) regulated by iron via 5′-IRE ALAS1 (liver) feedback-inhibited by haem and induced by CYP450-inducing drugs (→ precipitate acute porphyria). ALAS2 (bone marrow) regulated by iron via IRP/IRE — iron deficiency ↓ ALAS2 translation → ↓ haem synthesis.
🔴 Lead poisoning: inhibits BOTH ALA Dehydratase (Step 2) AND Ferrochelatase (Step 8) Results: ↑ urinary ALA (Step 2 block) + ↑ ZPP/FEP (Step 8 block — zinc substitutes for iron). Basophilic stippling on blood film is pathognomonic.
🔴 IDA: ↓ iron + ↑ TIBC + ↓ ferritin. ACD: ↓ iron + ↓ TIBC + ↑/normal ferritin TIBC (= transferrin) is the key differentiator: in IDA the body upregulates transferrin to capture iron (↑ TIBC); in ACD, inflammation suppresses transferrin synthesis (↓ TIBC).
🔴 HFE C282Y homozygous → ↓ hepcidin → ↑ ferroportin → iron overload Screening: fasting transferrin saturation >45% → check ferritin → HFE genotyping. Treatment: weekly phlebotomy until ferritin <50 ng/mL.
🟠 DcytB reduces Fe³⁺ → Fe²⁺ using ascorbate (Vitamin C). Hephaestin oxidises Fe²⁺ → Fe³⁺ using copper Iron changes oxidation state TWICE in the enterocyte: reduced apically for DMT1 uptake, re-oxidised basally for transferrin loading. The two enzymes explain Vitamin C and copper’s roles in iron absorption.
🟠 IRE/IRP system: TfR1 = 3′-IREs (stabilised by IRP when iron low); Ferritin = 5′-IRE (translation blocked by IRP when iron low) When iron is low, IRP maximises iron uptake (↑ TfR1) and minimises storage (↓ ferritin). When iron is high, the reverse. Reciprocal and elegant.
🟠 Reticulocyte count peaks 7–10 days after starting iron therapy — earliest sign of response Haemoglobin rises at 2–4 weeks (~1 g/dL/week). Continue 3–6 months post-normalisation to replenish stores.
🟠 Deferiprone = best cardiac iron clearance + weekly FBC mandatory (agranulocytosis risk) Deferasirox = most convenient oral (once daily, monthly renal monitoring). Deferoxamine = gold standard for severe overload (SC/IV infusion). Combination deferoxamine + deferiprone for severe cardiac iron loading.
🟡 Porphyria Cutanea Tarda (PCT) = Uroporphyrinogen Decarboxylase deficiency = most common porphyria overall Acute Intermittent Porphyria (AIP) = PBG Deaminase deficiency = most common acute porphyria (neuropsychiatric attacks, urine turns red-brown on standing, no cutaneous features).
🟡 Erythroferrone (ERFE) suppresses hepcidin during increased erythropoiesis EPO drives erythroblast proliferation → ERFE released → suppresses hepatic hepcidin → iron mobilised for haemoglobin. This explains why haemolytic anaemias and ineffective erythropoiesis (β-thalassaemia) lead to secondary iron overload despite NOT taking supplemental iron.
🟡 Plummer-Vinson (Paterson-Brown-Kelly) syndrome: IDA + oesophageal web + dysphagia — pre-malignant for post-cricoid carcinoma Middle-aged women; responds to iron treatment; requires endoscopy/dilatation.
🧠 Mnemonics
“SAL” Steps — Haem Synthesis Compartments Steps 1 = Mitochondria (ALAS — Succinyl-CoA + Glycine → ALA) Steps 2–5 = Cytoplasm (ALA → PBG → HMB → UPG-III → CPG-III) Steps 6–8 = Mitochondria (CPG-III → PPG-IX → PP-IX → Haem) Memory: “Starts in Mito → Leaves for Cyto → Returns to Mito”
ALAS → cytoplasm × 4 steps → back to mitochondria × 3 steps → Haem
“HALT-FLow” — When Hepcidin Rises (Iron Trapped) H = High iron stores A = Acute inflammation / infection (IL-6) L = Liver disease (advanced — but also produces hepcidin so complex) T = Transferrin saturation elevated
F = Fat (obesity → IL-6) L = LPS/bacterial products O = Overload (iron excess) w = With any inflammatory cytokine storm
Hepcidin FALLS with: Iron deficiency, Hypoxia, Haemolysis, Erythropoiesis (ERFE)
“DAH / BAS” — IDA vs ACD at a glance IDA: Down ferritin, Above-normal TIBC (↑), Hemoglobin microcytic ACD: Big ferritin (acute phase ↑), Abnormal TIBC (↓ — inflamed), Serum iron ↓ (trapped by hepcidin)
“5Bs of Haemochromatosis” Bronze skin · Bronze diabetes (pancreas) · Bad liver (cirrhosis/HCC) · Bad heart (cardiomyopathy) · Bare gonads (hypogonadism) + 2nd/3rd MCP joint arthritis (bonus B: “Bad joints”)
⚠️ Common Mistakes
❌ “TIBC is elevated in both IDA and ACD” ✅ TIBC is ↑ in IDA (more transferrin made to capture scarce iron) but ↓/normal in ACD (inflammation suppresses transferrin synthesis). TIBC is the key differentiator.
❌ “Serum ferritin is always reliable for iron stores” ✅ Ferritin is an acute-phase reactant. It can be falsely normal/elevated in IDA with concurrent infection, liver disease, or malignancy. Use sTfR index or bone marrow Prussian blue in equivocal cases.
❌ “Iron is absorbed as Fe³⁺ and exported as Fe²⁺” ✅ The reverse: Fe³⁺ is REDUCED to Fe²⁺ by DcytB (apical, for DMT1 uptake), then Fe²⁺ is OXIDISED back to Fe³⁺ by hephaestin (basolateral, for transferrin loading). Iron changes state twice during enterocyte transit.
❌ “ALAS is the rate-limiting enzyme and it only matters in liver” ✅ Two isoforms: ALAS1 (liver, haem-inhibited, drug-induced) and ALAS2 (erythroid, iron-regulated via IRE). ALAS2 mutations cause X-linked sideroblastic anaemia. Knowing both isoforms is essential.
❌ “Haemochromatosis is caused by too much ferritin” ✅ Haemochromatosis is caused by deficient hepcidin (from HFE/HJV/HAMP mutations) → ↑ ferroportin activity → excessive iron absorption. Ferritin overloads secondarily. The primary defect is in the hepcidin signalling cascade.
📝 5 Practice MCQs
Q1: A 7-year-old from a rural area has abdominal pain, intellectual delay and microcytic hypochromic anaemia. Blood film shows basophilic stippling. Urinary ALA is elevated. ZPP in RBCs is markedly raised. Which two haem synthesis enzymes are inhibited?
- A. ALAS and Ferrochelatase
- B. ALA Dehydratase and Ferrochelatase
- C. PBG Deaminase and Uroporphyrinogen Decarboxylase
- D. ALAS2 and Ferrochelatase
✅ Answer: B. ALA Dehydratase and Ferrochelatase
Lead poisoning. Basophilic stippling + ↑ urinary ALA + ↑ ZPP is the classic triad. Lead (Pb²⁺) inhibits ALA Dehydratase (Step 2 — displaces Zn²⁺ → ALA accumulates → ↑ urinary ALA) and Ferrochelatase (Step 8 — inhibits Fe²⁺ insertion → zinc substitutes → ZPP↑). Blood lead level confirms diagnosis. Treatment: chelation therapy (DMSA/succimer for children; EDTA for severe cases).
Q2: A 32-year-old woman with coeliac disease and Hb 8.6 g/dL, MCV 64 fL, serum iron low, TIBC elevated, ferritin 5 ng/mL has been taking oral ferrous sulphate for 3 months with no improvement. Best next step?
- A. Increase dose of oral iron + add Vitamin C
- B. Switch to IV iron (ferric carboxymaltose)
- C. Bone marrow biopsy to exclude haematological malignancy
- D. Add erythropoietin injections
✅ Answer: B. IV iron (ferric carboxymaltose)
Coeliac disease damages duodenal villi — the primary site of iron absorption. Oral iron cannot be absorbed regardless of dose. IV iron bypasses the GI tract and delivers iron directly to the bloodstream. This is the definitive management when oral iron fails due to malabsorption. EPO would be inappropriate (the problem is substrate, not EPO deficiency).
Q3: A 45-year-old man has fatigue, arthritis of 2nd/3rd MCP joints, skin bronzing, and family history of liver cirrhosis. Fasting transferrin saturation = 62%; ferritin = 1,840 ng/mL. Most likely genetic abnormality?
- A. MLH1 mutation (Lynch syndrome)
- B. HJV mutation (juvenile haemochromatosis)
- C. HFE C282Y homozygous mutation
- D. HAMP mutation causing hepcidin excess
✅ Answer: C. HFE C282Y homozygous mutation
Classic adult HFE haemochromatosis (Type 1) — C282Y homozygous in 85–90% of cases. Presentation: Bronze skin, MCP joint arthropathy (2nd/3rd fingers — characteristic), family history, markedly elevated transferrin saturation (>45% = screening threshold) and ferritin. C282Y disrupts HFE protein → impaired hepcidin signalling → excessive iron absorption. Treatment: weekly phlebotomy until ferritin <50 ng/mL. </details>
Q4: During iron absorption, dietary Fe³⁺ must be reduced before uptake. Which enzyme performs this reduction and what cofactor does it require?
- A. Hephaestin; copper
- B. DMT1; H⁺ gradient
- C. DcytB (Duodenal Cytochrome B); ascorbate (Vitamin C)
- D. Ferrochelatase; NADPH
✅ Answer: C. DcytB; ascorbate (Vitamin C)
DcytB is the apical brush-border ferrireductase that reduces Fe³⁺ → Fe²⁺ using ascorbate as the electron donor. This is the molecular basis for Vitamin C enhancing iron absorption. DMT1 then transports Fe²⁺ into the cell (it cannot transport Fe³⁺). Hephaestin performs the REVERSE reaction (Fe²⁺→Fe³⁺) at the basolateral membrane using copper. Ferrochelatase inserts Fe²⁺ into protoporphyrin (haem synthesis, not absorption).
Q5: A thalassaemia major patient on chronic transfusions develops cardiomyopathy. Ferritin is 4,200 ng/mL. Which chelator has best cardiac iron clearance and requires weekly blood monitoring?
- A. Deferoxamine; monthly spirometry
- B. Deferasirox; monthly creatinine
- C. Deferiprone; weekly FBC (agranulocytosis risk)
- D. Deferoxamine; weekly audiometry
✅ Answer: C. Deferiprone; weekly FBC
Deferiprone is a small lipophilic bidentate chelator that uniquely penetrates cell membranes → best cardiac iron clearance. Its serious adverse effect is agranulocytosis (~1–2% of patients) — requiring mandatory weekly full blood count. If neutrophils fall below 1.5×10⁹/L, stop immediately. Combination deferoxamine + deferiprone is used for severe cardiac iron loading (synergistic chelation). Deferasirox (monthly creatinine) is best for general overload monitoring but has poor cardiac penetration.
📚 References
📖 Harper’s Illustrated Biochemistry — Chapter 31: Porphyrins & Bile Pigments; Chapter 52: Iron section 📖 Lippincott’s Illustrated Reviews: Biochemistry — Chapter 28: Vitamins and Minerals (Iron) 📖 Williams Haematology — Chapter 42: Disorders of Iron Metabolism 📖 Harrison’s Principles of Internal Medicine — Chapter 428: Iron Deficiency and Other Hypoproliferative Anaemias 📖 Guyton & Hall Medical Physiology — Chapter 33: RBCs, Anaemia and Polycythaemia
🚀 Keep Practising
Iron metabolism questions in NEET PG and USMLE integrate biochemistry, physiology, pathology, and pharmacology into single clinical scenarios. The story: iron enters as Fe²⁺ (DcytB + DMT1) → exported as Fe³⁺ (hephaestin + ferroportin) → carried by transferrin to bone marrow → incorporated into haem (ferrochelatase) → regulated by hepcidin. When any step breaks, you predict the consequence.