What You Will Learn
- Draw Haworth and Fischer projections of glucose, fructose, galactose; understand D/L, α/β, reducing/non-reducing sugars
- Trace all 10 glycolytic steps with enzymes, cofactors, and regulation
- Explain gluconeogenesis — the 4 unique bypass enzymes and their regulation
- Describe the pentose phosphate pathway — oxidative and non-oxidative phases
- Map fructose and galactose metabolism with their inherited enzyme deficiency disorders
- Identify clinical disorders: galactosaemia, fructosaemia, glycogen storage diseases, diabetes mellitus, G6PD deficiency
- Apply HbA1c, sorbitol pathway, and AGEs to diabetic complications
Section 1 — Carbohydrate Chemistry
1.1 Classification
| Class | Definition | Examples |
|---|---|---|
| Monosaccharides | Single unit; cannot be hydrolysed | Glucose, Fructose, Galactose, Ribose |
| Disaccharides | Two monosaccharides + glycosidic bond | Lactose, Sucrose, Maltose |
| Oligosaccharides | 3–10 units | Raffinose, Stachyose |
| Polysaccharides | >10 units | Starch, Glycogen, Cellulose |
1.2 Glucose — The Central Sugar
D-Glucose: Aldohexose (C₆H₁₂O₆); aldehyde at C1; D-configuration (OH on right at C5 in Fischer projection)
D vs L: Determined by configuration at C5 (for hexoses). Nearly all biological sugars are D-configuration.
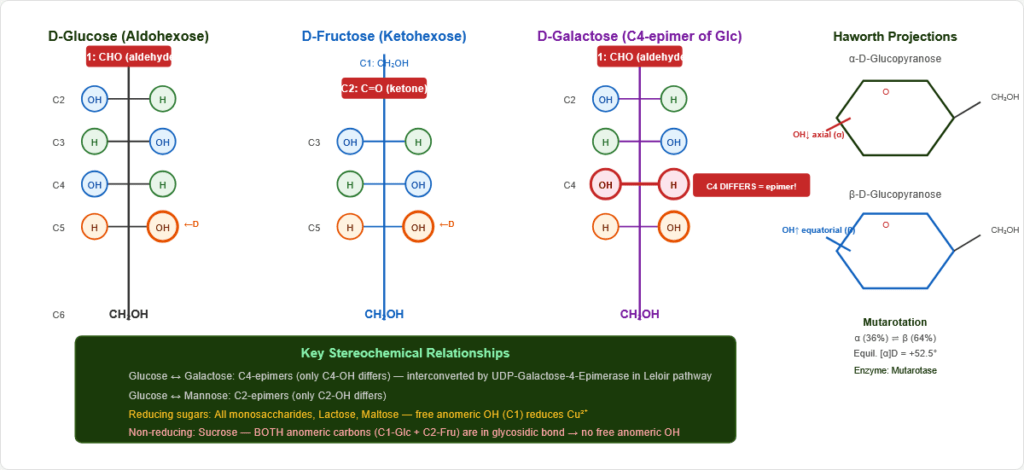
Figure 2: Fischer projection of D-glucose. The aldehyde at C1 and D-configuration at C5 (OH on right) define this molecule. All four chiral centres contribute to its unique stereochemistry.
1.3 Ring Formation — Haworth Projections and Anomers
The C1 aldehyde reacts with C5-OH → pyranose ring (six-membered):
- α-D-Glucopyranose: C1-OH axial (below ring plane) — same side as ring oxygen
- β-D-Glucopyranose: C1-OH equatorial (above ring plane) — thermodynamically more stable
Mutarotation: α ⇌ open chain ⇌ β in solution; equilibrium = 36% α + 64% β (specific rotation +52.5°). Enzyme: Mutarotase (aldose-1-epimerase)
1.4 Key Relationships Between Sugars
Epimers: Differ at ONE non-anomeric carbon
- Glucose ↔ Galactose: C4 epimers
- Glucose ↔ Mannose: C2 epimers
Reducing sugars: Free anomeric C1-OH → can reduce Cu²⁺ (Benedict’s/Fehling’s)
- Reducing: All monosaccharides, Lactose, Maltose
- Non-reducing: Sucrose (BOTH anomeric carbons locked in glycosidic bond)
1.5 Clinically Important Sugars
| Sugar | Class | Key Role | Clinical Significance |
|---|---|---|---|
| D-Glucose | Aldohexose | Universal fuel; brain’s obligate substrate | Diabetes mellitus; hypoglycaemia |
| D-Fructose | Ketohexose (C2 ketone) | Hepatic metabolism → DHAP/G3P | Hereditary fructose intolerance |
| D-Galactose | C4 epimer of glucose | Leloir pathway → Glc-6-P | Classic galactosaemia; cataracts |
| D-Ribose | Aldopentose | RNA backbone; ATP, NAD⁺, CoA | Pentose phosphate pathway product |
| D-2-Deoxyribose | Deoxyaldopentose | DNA backbone | PPP → ribonucleotide reductase |
| D-Mannose | C2 epimer of glucose | N-glycosylation; Man-6-P lysosomal targeting | I-cell disease |
| L-Fucose | Deoxyhexose | ABO blood group antigens | Blood group chemistry |
1.6 Disaccharides and Lactose Intolerance
| Disaccharide | Components | Bond | Digestive Enzyme | Deficiency |
|---|---|---|---|---|
| Lactose (milk sugar) | Galactose + Glucose | β-1,4 | Lactase (brush border) | Lactose intolerance: osmotic diarrhoea + bloating |
| Sucrose (table sugar) | Glucose + Fructose | α,β-1,2 (non-reducing) | Sucrase | Rare |
| Maltose | Glucose + Glucose | α-1,4 | Maltase | Rare |
| Trehalose | Glucose + Glucose | α,α-1,1 (non-reducing) | Trehalase | Rare |
Lactose intolerance: Lactase deficiency → undigested lactose → osmotic diarrhoea + bacterial fermentation → gas (H₂, CO₂) + short-chain fatty acids → bloating, flatulence.
- Primary: Genetic; common in Asians (~90%), Africans, Middle Easterners; European lactase persistence (LCT gene polymorphism) is the exception
- Diagnosis: Hydrogen breath test (>20 ppm rise after lactose load); lactose tolerance test
Section 2 — Glycolysis: 10 Steps in the Cytoplasm
2.1 Overview
Converts glucose (6C) → 2 pyruvate (3C); NET: +2 ATP + 2 NADH
- Location: Cytoplasm (all 10 steps)
- Functions in aerobic and anaerobic conditions
- Irreversible steps at 1, 3, 10 (Hexokinase/Glucokinase, PFK-1, Pyruvate Kinase)
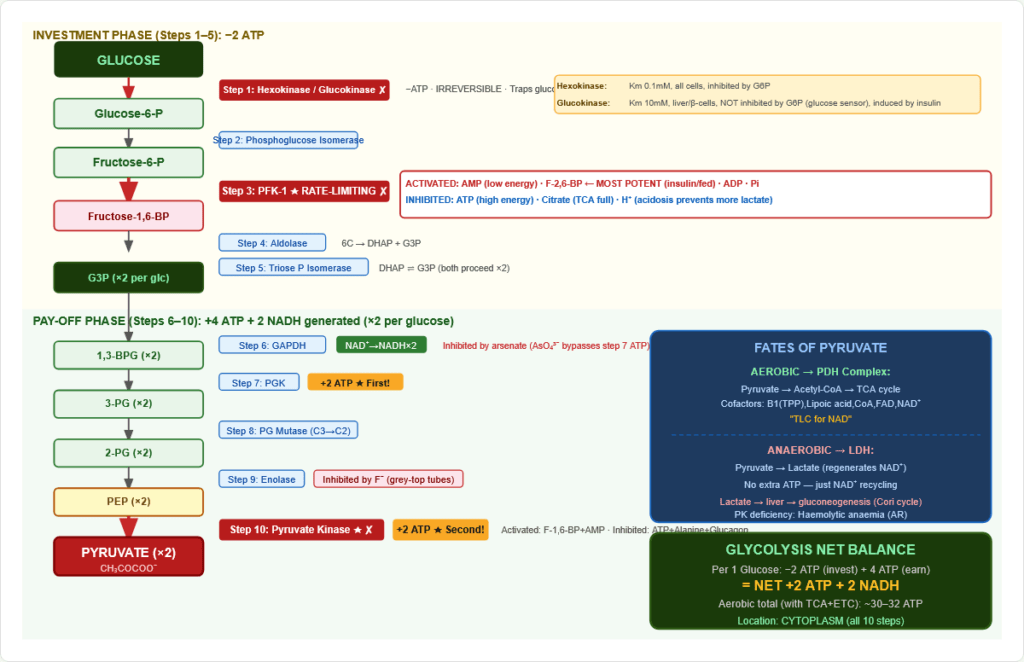
Figure 4: Complete glycolysis pathway. Investment phase (steps 1–5): 2 ATP consumed. Pay-off phase (steps 6–10): 4 ATP + 2 NADH generated. Net: +2 ATP +2 NADH per glucose. Three irreversible steps: Hexokinase (step 1), PFK-1 (step 3), Pyruvate Kinase (step 10).
2.2 The 10 Steps
INVESTMENT PHASE (Steps 1–5) — 2 ATP consumed
Step 1: Glucose → Glucose-6-Phosphate (G6P)
- Enzyme: Hexokinase (all cells) / Glucokinase (liver, β-cells)
- Cofactor: ATP + Mg²⁺; Irreversible; Traps glucose in cell
| Property | Hexokinase | Glucokinase (Hexokinase IV) |
|---|---|---|
| Km for glucose | ~0.1 mM (HIGH affinity) | ~10 mM (LOW affinity) |
| Inhibition by G6P | YES (product inhibition) | NO |
| Tissue | All cells | Liver, Pancreatic β-cells |
| Regulation | Constitutive | Induced by insulin |
| Function | Works at all glucose levels | Glucose sensor — only active at high glucose |
Step 2: G6P → Fructose-6-Phosphate (F6P)
- Enzyme: Phosphoglucose isomerase (PGI); Reversible; Aldose → ketose
Step 3: F6P → Fructose-1,6-bisphosphate (F-1,6-BP) ★ RATE-LIMITING
- Enzyme: Phosphofructokinase-1 (PFK-1) — THE rate-limiting enzyme of glycolysis
- Cofactor: ATP + Mg²⁺; Irreversible — committed step
Regulation of PFK-1 (most important glycolytic regulation):
| Activators | Inhibitors |
|---|---|
| AMP ↑ (low energy) | ATP ↑ (high energy) |
| Fructose-2,6-bisphosphate (F-2,6-BP) — MOST POTENT activator | Citrate (TCA full) |
| ADP ↑, Pi ↑ | H⁺ / acidosis |
F-2,6-BP: Made by PFK-2 (active in fed state, insulin-activated). NOT a glycolytic intermediate — purely regulatory. Simultaneously inhibits FBPase-1 → ensures glycolysis and gluconeogenesis don’t run together.
Step 4: F-1,6-BP → DHAP + Glyceraldehyde-3-Phosphate (G3P)
- Enzyme: Aldolase; Cleaves 6C → two 3C trioses
Step 5: DHAP → G3P
- Enzyme: Triose Phosphate Isomerase (TPI); Both trioses now proceed as G3P
- TPI deficiency: Very rare; haemolytic anaemia + neuropathy; DHAP accumulation toxic
PAY-OFF PHASE (Steps 6–10) — ×2 per glucose
Step 6: G3P → 1,3-Bisphosphoglycerate (1,3-BPG)
- Enzyme: GAPDH (Glyceraldehyde-3-phosphate dehydrogenase)
- Cofactor: NAD⁺ → NADH (first NADH generation)
- Inhibited by arsenate (AsO₄³⁻) → bypasses step 7 ATP generation → arsenate poisoning
Step 7: 1,3-BPG → 3-Phosphoglycerate (3-PG)
- Enzyme: Phosphoglycerate kinase (PGK)
- First ATP generation (×2 = 2 ATP per glucose; substrate-level phosphorylation)
Step 8: 3-PG → 2-Phosphoglycerate (2-PG)
- Enzyme: Phosphoglycerate mutase; Moves phosphate C3 → C2
Step 9: 2-PG → Phosphoenolpyruvate (PEP)
- Enzyme: Enolase; Dehydration → high-energy enol phosphate
- Inhibited by fluoride (F⁻) — used in grey-top blood tubes (NaF) to preserve glucose in blood samples
Step 10: PEP → Pyruvate ★ IRREVERSIBLE
- Enzyme: Pyruvate Kinase (PK)
- Second ATP generation (×2 = 2 ATP per glucose)
- Activated by: F-1,6-BP (feedforward), AMP
- Inhibited by: ATP, Alanine, Glucagon (phosphorylates PK in liver → inactive)
- PK deficiency (AR): Most common glycolytic enzyme deficiency; haemolytic anaemia (RBCs depend entirely on glycolysis); ↑ 2,3-BPG (partially compensates)
2.3 Net Glycolytic Balance
| Per glucose | |
|---|---|
| ATP consumed (steps 1, 3) | −2 |
| ATP generated (steps 7, 10, ×2 each) | +4 |
| Net ATP | +2 |
| Net NADH | +2 (step 6, ×2) |
| Net Pyruvate | 2 molecules |
2.4 Fates of Pyruvate
Aerobic: Pyruvate → Acetyl-CoA (PDH complex) → TCA cycle → ~28–30 ATP more Anaerobic: Pyruvate → Lactate (LDH) — regenerates NAD⁺ for continued GAPDH activity
Pyruvate Dehydrogenase Complex (PDH):
- Location: Mitochondrial matrix
- Cofactors: “TLC for NAD” — Thiamine-PP (B1), Lipoic acid, CoA (B5), FAD (B2), NAD⁺ (B3)
- Regulation:
- PDH Kinase (activated by NADH, Acetyl-CoA, ATP) → phosphorylates E1 → INACTIVATES PDH
- PDH Phosphatase (activated by Ca²⁺, insulin) → dephosphorylates → ACTIVATES PDH
- PDH deficiency (X-linked): Lactic acidosis from birth; elevated pyruvate and lactate; neurological damage; treat with ketogenic diet + thiamine
2.5 Total ATP from Complete Glucose Oxidation
| Stage | ATP |
|---|---|
| Glycolysis (substrate level) | 2 |
| Glycolytic NADH (×2) via ETC | 5 (×2.5/NADH) |
| PDH NADH (×2) via ETC | 5 |
| TCA NADH (×6) via ETC | 15 |
| TCA FADH₂ (×2) via ETC | 3 (×1.5/FADH₂) |
| TCA GTP (×2) | 2 |
| TOTAL | ~30–32 ATP |
Section 3 — Gluconeogenesis: Synthesising Glucose from Non-Carbohydrates
3.1 Overview
- Location: Liver (90%); Kidney cortex (10%, more important in prolonged starvation/acidosis)
- Substrates: Lactate (Cori cycle), Amino acids (alanine, glutamine), Glycerol (from lipolysis), Propionate (odd-chain FA → propionyl-CoA → succinyl-CoA → OAA)
- NOT from Acetyl-CoA: Even-chain fatty acids CANNOT be converted to glucose net (no glyoxylate cycle in humans)
3.2 The 4 Unique Bypass Enzymes
Gluconeogenesis reverses glycolysis EXCEPT at 3 irreversible steps — requiring 4 unique enzymes:
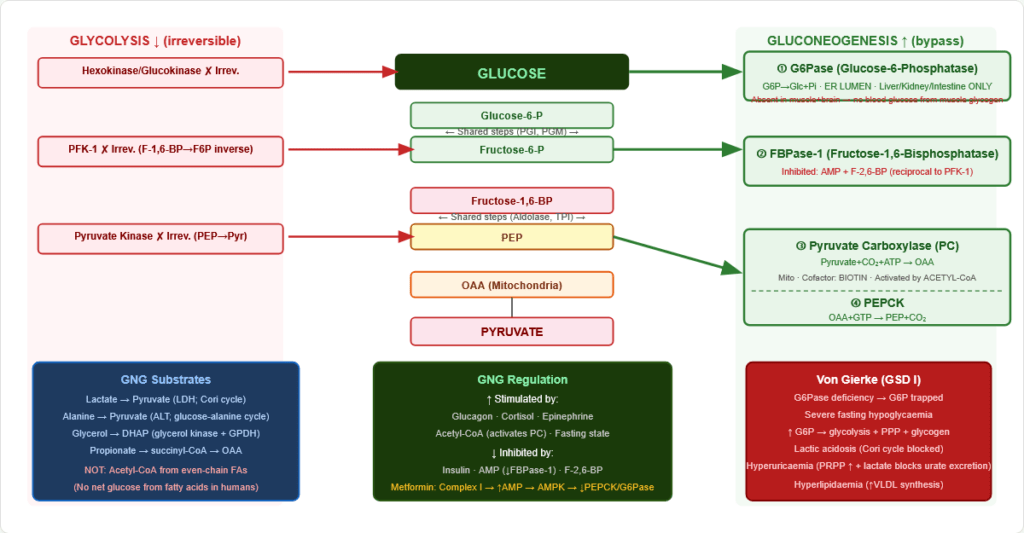
Figure 5: Gluconeogenesis bypass enzymes. The four unique enzymes circumvent the three irreversible glycolytic reactions. “Please Don’t Forget Glucose” mnemonic: Pyruvate Carboxylase, (don’t skip) PEPCK, Fructose-1,6-Bisphosphatase, Glucose-6-Phosphatase.
Bypass 1 — Pyruvate → PEP (bypasses Pyruvate Kinase):
- Pyruvate Carboxylase (PC): Pyruvate + CO₂ + ATP → OAA
- Location: Mitochondria
- Cofactor: Biotin (CO₂ carrier)
- Activated by: Acetyl-CoA (signals fat oxidation → time to make glucose)
- PC deficiency → lactic acidosis + hyperammonaemia + hypoglycaemia
- PEPCK (Phosphoenolpyruvate Carboxykinase): OAA + GTP → PEP + CO₂
- Location: Cytoplasm (mainly); also mitochondria
- Induced by: Glucagon, cortisol, thyroid hormones
- Inhibited by: Insulin
- Metformin mechanism: Inhibits Complex I → ↑ AMP → AMPK → ↓ CRTC2 → ↓ PEPCK + G6Pase transcription → ↓ hepatic gluconeogenesis
Bypass 2 — F-1,6-BP → F6P (bypasses PFK-1):
- Fructose-1,6-Bisphosphatase (FBPase-1)
- Inhibited by: AMP, F-2,6-BP (reciprocal to PFK-1)
- Activated by: Citrate
Bypass 3 — G6P → Glucose (bypasses Hexokinase):
- Glucose-6-Phosphatase (G6Pase)
- Location: ER lumen (unique!)
- Present in: Liver, kidney, intestine — NOT in muscle or brain
- Von Gierke disease (GSD Type I): G6Pase deficiency → trapped G6P → severe fasting hypoglycaemia + hepatomegaly + lactic acidosis + hyperuricaemia + hyperlipidaemia
3.3 Gluconeogenesis Regulation
Activated by (fasting state): Glucagon (↑ PEPCK, ↓ PFK-2 → ↓ F-2,6-BP, ↑ FBPase-1), Cortisol (induces PEPCK), Epinephrine, Acetyl-CoA (activates PC)
Inhibited by (fed state): Insulin (↓ PEPCK, ↑ PFK-2 → ↑ F-2,6-BP), AMP (↓ FBPase-1)
Section 4 — The Pentose Phosphate Pathway (PPP)
4.1 Overview
- Location: Cytoplasm
- Products: NADPH + Ribose-5-phosphate
- Main tissues: Liver (FA synthesis), RBCs (antioxidant), adrenal cortex (steroidogenesis), mammary gland, gonads, leukocytes
4.2 Oxidative Phase (Irreversible) — Generates NADPH
Step 1: G6P → 6-Phosphogluconolactone + NADPH
- Enzyme: Glucose-6-Phosphate Dehydrogenase (G6PD) — RATE-LIMITING enzyme of PPP
- Cofactor: NADP⁺ (NOT NAD⁺)
Step 2: 6-Phosphogluconolactone → 6-Phosphogluconate
- Enzyme: Lactonase
Step 3: 6-Phosphogluconate → Ribulose-5-P + CO₂ + NADPH
- Enzyme: 6-Phosphogluconate dehydrogenase
- Second NADPH; CO₂ released (irreversibility)
G6PD Deficiency — Most common human enzymopathy (400 million affected; X-linked recessive):
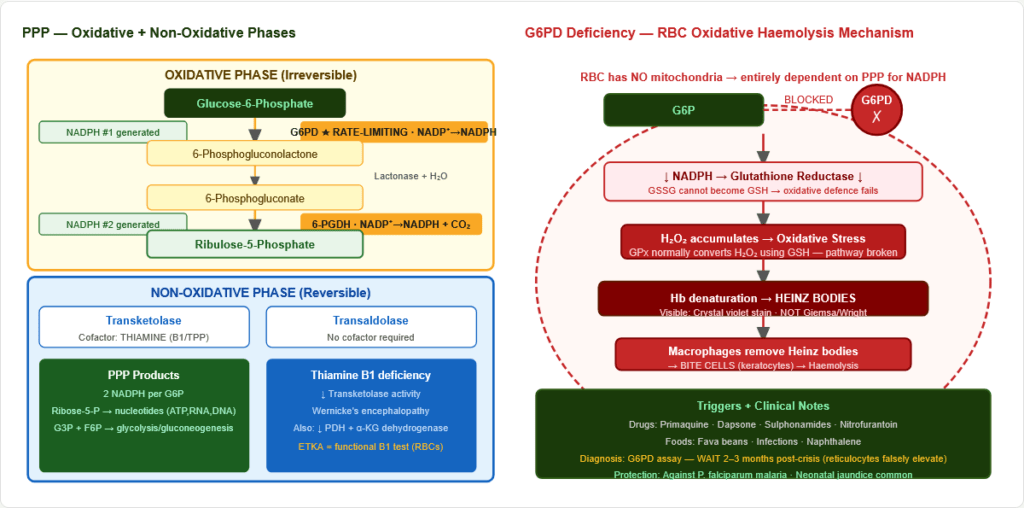
Figure 6: G6PD deficiency mechanism. RBCs have no mitochondria and rely entirely on PPP for NADPH. G6PD deficiency → NADPH depletion → GSH cannot be regenerated → oxidative stress → Heinz bodies (denatured Hb) → bite cells (macrophage removal of Heinz bodies) → haemolytic anaemia.
- RBCs: No mitochondria → entirely dependent on PPP for NADPH → glutathione (GSH) cannot be regenerated → oxidative haemolysis
- Triggers: Primaquine, dapsone, sulphonamides, fava beans, infections, naphthalene
- Blood film: Heinz bodies (crystal violet stain) + Bite cells
- Neonatal jaundice: Impaired conjugation (bilirubin → water-soluble) — requires NADPH
- Diagnosis: G6PD enzyme assay — perform 2–3 months AFTER crisis (reticulocytes have higher G6PD → false-normal during haemolysis)
- Malaria protection: P. falciparum preferentially infects normal RBCs; G6PD-deficient RBCs are hostile
4.3 Non-Oxidative Phase (Reversible)
Interconverts pentose phosphates with glycolytic intermediates:
- Transketolase (cofactor: Thiamine/B1 as TPP) — functional thiamine deficiency test (ETKA in RBCs)
- Transaldolase (no cofactor)
Wernicke’s encephalopathy (B1 deficiency): Impairs Transketolase (PPP), PDH, and α-ketoglutarate dehydrogenase (TCA) → encephalopathy + ataxia + ophthalmoplegia.
Section 5 — Fructose Metabolism
5.1 Normal Pathway in Hepatocytes
Figure 7: Hepatic fructose metabolism. Fructose bypasses PFK-1 (the rate-limiting step of glycolysis) → enters as DHAP and G3P unregulated → can be converted to fat more readily than glucose. Fructokinase deficiency = benign; Aldolase B deficiency = HFI (toxic F1P accumulation).
Step 1: Fructose → Fructose-1-Phosphate (F1P) by Fructokinase
- Bypasses PFK-1 regulation → fructose enters glycolysis unregulated → contributes to NAFLD and hypertriglyceridaemia with high fructose consumption
Step 2: F1P → DHAP + Glyceraldehyde by Aldolase B (liver-specific)
Step 3: Glyceraldehyde → G3P by Triokinase
5.2 Disorders of Fructose Metabolism
Essential Fructosuria (Fructokinase deficiency):
- Benign; no symptoms; fructose accumulates in blood + urine
- Urinalysis: Clinitest positive (reducing sugar) but glucose oxidase dipstick NEGATIVE
- No treatment needed
Hereditary Fructose Intolerance (HFI) — Aldolase B deficiency:
- F1P accumulates → TOXIC: inhibits glycogen phosphorylase + gluconeogenesis → severe hypoglycaemia; depletes Pi → depletes ATP → liver damage
- Clinical: Symptoms after weaning (sucrose/fructose introduction); vomiting, hypoglycaemia, liver failure, Fanconi syndrome (renal tubular dysfunction)
- Pathognomonic: Self-aversion to sweet foods develops (life-saving self-protection!)
- Treatment: Eliminate ALL fructose, sucrose, and sorbitol from diet
- Lab: ↑ F1P; ↑ ALT/AST; hypoglycaemia after fructose; ↑ urine reducing substances
Section 6 — Galactose Metabolism: The Leloir Pathway
6.1 Four-Step Pathway (Galactose → Glucose-6-P)
| Step | Enzyme | Deficiency | Disease | Consequence |
|---|---|---|---|---|
| 1 | Galactokinase | Galactokinase deficiency | Galactosaemia Type II | Galactose + galactitol accumulate → cataracts ONLY; no liver damage |
| 2 | GALT (Galactose-1-P Uridylyltransferase) | Classic galactosaemia | Type I (most severe) | Gal-1-P (TOXIC); liver failure, cataracts, E. coli sepsis, ovarian failure |
| 3 | UDP-Galactose-4-Epimerase | GALE deficiency | Type III | Variable; can be benign |
| 4 | Phosphoglucomutase | — | — | — |
6.2 Classic Galactosaemia (GALT Deficiency) — Type I
Inheritance: Autosomal recessive Toxicity: Gal-1-P inhibits phosphoglucomutase → blocks glycogen metabolism; galactose → galactitol in non-liver tissues (aldose reductase)
Clinical triad after milk feeding:
- Jaundice + liver failure (direct hyperbilirubinaemia, hepatomegaly → cirrhosis)
- Cataracts (galactitol osmotic damage to lens)
- E. coli neonatal septicaemia (Gal-1-P impairs neutrophil bactericidal function — PATHOGNOMONIC)
Other features: Hypoglycaemia, Fanconi syndrome (aminoaciduria, phosphaturia), intellectual disability if untreated
Long-term complications (even with treatment):
- Ovarian failure (primary ovarian insufficiency) in >80% of females — galactitol damage
- Speech/language delays; ataxia
Diagnosis: Beutler test (GALT enzyme activity in blood spot); ↑ Gal-1-P in RBCs; urine reducing substances positive
Treatment: Eliminate ALL galactose and lactose (use soy-based formula from birth)
Section 7 — Blood Glucose Regulation and Diabetes
7.1 Normal Glucose Homeostasis
- Fasting: 70–100 mg/dL (3.9–5.6 mmol/L)
- 2-hour postprandial: <140 mg/dL
Counter-regulatory hormones (raise glucose): Glucagon → glycogenolysis + gluconeogenesis + ketogenesis; Cortisol → gluconeogenesis + anti-insulin; Epinephrine → glycogenolysis + lipolysis; GH → anti-insulin (diabetogenic)
Glucose-lowering: Insulin (master; GLUT4 translocation, glycogen synthesis, FA synthesis, anti-gluconeogenesis); GLP-1 (incretin — glucose-dependent insulin secretion)
7.2 Insulin Secretion — KATP Channel Mechanism
Glucose-stimulated insulin secretion (GSIS):
- Glucose → β-cell via GLUT2
- Glucokinase phosphorylates → G6P → glycolysis → ↑ ATP:ADP ratio
- K_ATP channels (SUR1/Kir6.2) close → membrane depolarises
- Voltage-gated Ca²⁺ channels open → Ca²⁺ influx
- Exocytosis of insulin granules
Drug targets:
- Sulfonylureas (glibenclamide, glipizide): Close K_ATP → depolarise → insulin release (insulin secretagogues; hypoglycaemia risk — glucose-independent)
- GLP-1 agonists (liraglutide, semaglutide): Glucose-dependent insulin secretion (minimal hypoglycaemia)
- DPP-4 inhibitors (sitagliptin): Prolong GLP-1 → incretin effect
7.3 Diabetes Classification
| Type | Mechanism | Key Feature |
|---|---|---|
| Type 1 DM | Autoimmune β-cell destruction → absolute insulin deficiency | Anti-GAD, anti-islet antibodies; DKA-prone; any age |
| Type 2 DM | Insulin resistance + progressive β-cell dysfunction | Obesity; strong genetic component; usually >40 |
| MODY 2 (GCK) | Glucokinase mutation → raised glucose set point | Mild stable hyperglycaemia; no progression; no treatment usually |
| MODY 3 (HNF1A) | HNF1α mutation → progressive β-cell failure | Most common MODY; responds dramatically to low-dose sulfonylurea |
| Gestational DM | Placental HPL → insulin resistance | Develops during pregnancy; OGTT diagnosis |
| LADA | Slow autoimmune (Type 1 variant) | Anti-GAD positive; initially misdiagnosed as Type 2 |
7.4 Diabetic Ketoacidosis (DKA) Biochemistry
Cascade: ↓ Insulin → ↑ Lipolysis → ↑ FFA → liver β-oxidation → ↑ Acetyl-CoA → Ketogenesis:
- Acetyl-CoA → Acetoacetate → β-Hydroxybutyrate (main ketone; reduced form, not detected by nitroprusside strips)
- Acetoacetate → Acetone (spontaneous; “fruity breath”)
DKA biochemical features:
- Blood glucose >250 mg/dL
- pH <7.3; HCO₃⁻ <15 mEq/L
- Anion gap elevated (>20; normal 8–12)
- Serum K⁺ initially high (acidosis shifts K⁺ out of cells) — total body K⁺ depleted
7.5 HbA1c — Glycated Haemoglobin
Formation: Glucose + Hb β-chain N-terminal valine → Schiff base (reversible aldimine) → Amadori rearrangement → HbA1c (irreversible ketoamine)
Reflects average blood glucose over 2–3 months (RBC lifespan ~120 days)
| HbA1c | Interpretation |
|---|---|
| <5.7% | Normal |
| 5.7–6.4% | Prediabetes |
| ≥6.5% | Diabetes (diagnostic cut-off) |
| Target for most patients | <7% (<53 mmol/mol) |
Falsely LOW: Haemolytic anaemia (↓ RBC lifespan → less glycation time), blood loss, haemoglobinopathies (HbS, HbC, HbE), pregnancy, iron deficiency treatment (new RBCs dilute old glycated ones)
Falsely HIGH: Iron deficiency anaemia (↓ new RBC production → older RBCs → more glycation time), B12/folate deficiency, splenectomy (↑ RBC lifespan)
7.6 The Sorbitol (Polyol) Pathway — Diabetic Complications
In hyperglycaemia, insulin-independent tissues (nerve, lens, kidney, blood vessels) have excess intracellular glucose → polyol pathway activated:
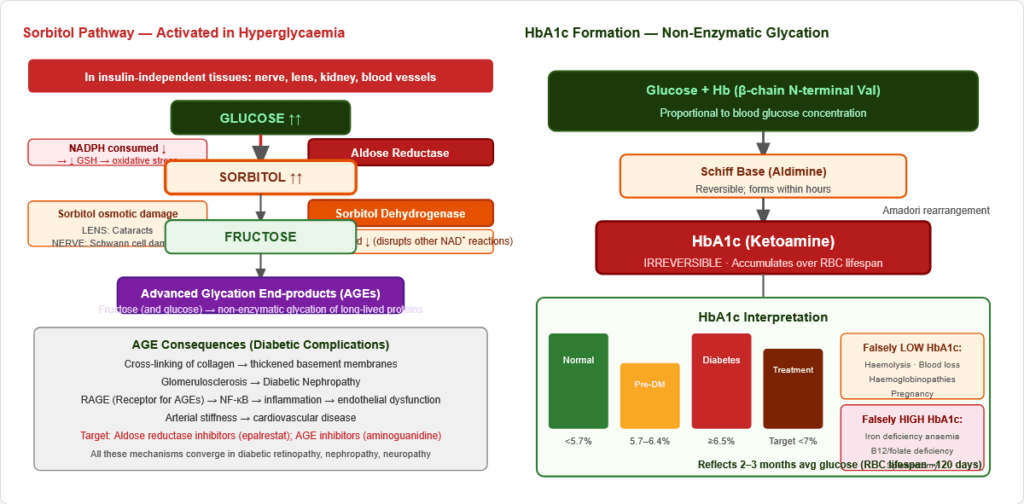
Figure 9: The sorbitol (polyol) pathway activated in hyperglycaemia. Aldose reductase (NADPH-dependent) converts glucose to sorbitol. Consequences: (1) sorbitol osmotic damage; (2) NADPH depletion → oxidative stress; (3) NAD⁺ consumption disrupts other reactions. Target for aldose reductase inhibitors (epalrestat).
Glucose → Sorbitol: Aldose Reductase (NADPH consumed) Sorbitol → Fructose: Sorbitol Dehydrogenase (NAD⁺ consumed)
Complications:
- Lens: Cataracts (sorbitol → osmotic damage)
- Peripheral nerves: Schwann cell damage → neuropathy
- Retinal pericytes: Retinopathy
- Kidney: Nephropathy contribution
Section 8 — Glycogen Storage Diseases (GSDs)
8.1 Glycogen Structure
- Glucose polymer: α-1,4 bonds (straight chain) + α-1,6 bonds (branch points every 8–12 residues)
- Liver glycogen (~100g): Maintains blood glucose during fasting
- Muscle glycogen (~400g): Local fuel only (no G6Pase → cannot release free glucose)
8.2 GSD Summary Table
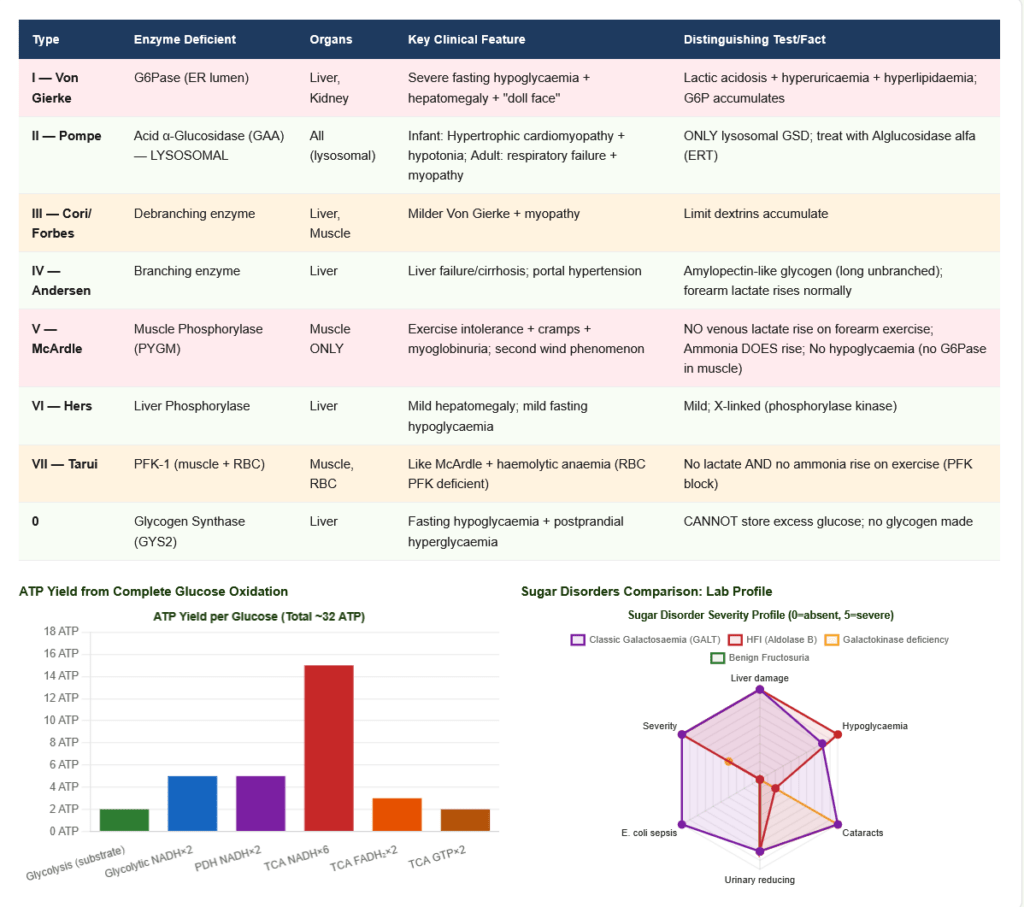
Show Image Figure 10: Glycogen storage disease pathway map. Each GSD type corresponds to a specific enzyme defect in glycogen synthesis or degradation. The clinical presentation depends on which tissues express the defective enzyme and what substrate accumulates.
| Type | Enzyme Deficient | Organs | Key Clinical Feature | Key Lab/Test |
|---|---|---|---|---|
| I — Von Gierke | G6Pase (ER lumen) | Liver, Kidney | Severe fasting hypoglycaemia + hepatomegaly + “doll-face” + lactic acidosis + hyperuricaemia + hyperlipidaemia | G6P accumulates; no free glucose release |
| II — Pompe | Acid α-Glucosidase (GAA) — Lysosomal | ALL (lysosomal) | Infantile: hypertrophic cardiomyopathy + hypotonia + CK↑↑; Adult: respiratory failure + myopathy | ONLY GSD with lysosomal enzyme; treat with alglucosidase alfa (ERT) |
| III — Cori/Forbes | Debranching enzyme | Liver, Muscle | Milder Von Gierke; myopathy | Limit dextrins accumulate |
| IV — Andersen | Branching enzyme | Liver | Liver failure/cirrhosis | Amylopectin-like glycogen (abnormal, long unbranched chains) |
| V — McArdle | Muscle Phosphorylase (PYGM) | Muscle ONLY | Exercise cramps + myoglobinuria; NO venous lactate rise on forearm exercise test; second wind phenomenon | CK markedly elevated; No lactate rise but ammonia DOES rise (distinguishes from PFK deficiency) |
| VI — Hers | Liver Phosphorylase | Liver | Mild hepatomegaly; mild hypoglycaemia | — |
| VII — Tarui | PFK-1 (muscle + RBC) | Muscle, RBC | Like McArdle + haemolytic anaemia (RBC PFK also deficient) | No lactate AND no ammonia rise (PFK block prevents AMP cycling) |
| 0 | Glycogen Synthase (GYS2) | Liver | Fasting hypoglycaemia + postprandial hyperglycaemia (cannot store excess) | No glycogen made |
Forearm ischaemic exercise test:
- Normal: Lactate ↑ AND Ammonia ↑ (glycolysis + purine nucleotide cycle)
- McArdle (Type V): Lactate does NOT rise; Ammonia DOES rise
- Tarui (Type VII): Lactate does NOT rise; Ammonia does NOT rise (PFK block disables both)
Section 9 — Connections
→ TCA Cycle: Pyruvate (from glycolysis) → Acetyl-CoA (PDH) → TCA. Gluconeogenesis uses OAA from TCA. Citrate exits mitochondria → Acetyl-CoA (for FA synthesis) + OAA (back to malate for gluconeogenesis). The metabolic axes are intimately coupled.
→ Lipid Metabolism: Excess glucose → Acetyl-CoA → de novo lipogenesis. Malonyl-CoA (from ACC, activated by insulin) inhibits CPT-1 → prevents FA oxidation in the fed state. Starvation: FA oxidation provides Acetyl-CoA → ketogenesis. Glycerol-3-P from DHAP (glycolysis) is the triglyceride backbone.
→ Amino Acid Metabolism: Glucose-alanine cycle: Muscle → pyruvate + glutamate → ALT → alanine → liver → pyruvate → gluconeogenesis + urea cycle for the nitrogen. Most glucogenic amino acids enter as OAA, pyruvate, α-ketoglutarate, or succinyl-CoA.
→ Nucleotide Synthesis: PPP provides ribose-5-P for purine + pyrimidine synthesis. NADPH from PPP drives ribonucleotide reductase. Rapidly dividing cells divert glucose to PPP.
→ Haemoglobin: 2,3-BPG (from Rapoport-Luebering shunt: 1,3-BPG → 2,3-BPG → 3-PG, operating only in RBCs) stabilises deoxy-Hb → right-shifts O₂ dissociation curve → ↑ O₂ delivery to tissues. High altitude: ↑ 2,3-BPG → adaptation. PK deficiency: upstream intermediates including 2,3-BPG accumulate → right-shift → partially compensates for anaemia.
→ Antioxidant Defence: NADPH from PPP → glutathione reductase → GSH regeneration. G6PD deficiency breaks this chain → oxidative haemolysis. Liver NADPH also powers cytochrome P450 monooxygenases.
High-Yield Exam Facts
🔴 PFK-1 = rate-limiting enzyme of glycolysis; F-2,6-BP = most potent activator; simultaneously inhibits FBPase-1 F-2,6-BP is made by PFK-2 (activated by insulin). Glucagon → PKA → inhibits PFK-2 → ↓ F-2,6-BP → ↓ glycolysis + ↑ gluconeogenesis. Insulin → activates PFK-2 → ↑ F-2,6-BP → ↑ glycolysis + ↓ gluconeogenesis. F-2,6-BP is NOT a glycolytic intermediate.
🔴 G6Pase = ER lumen; absent in muscle and brain → muscle glycogen cannot raise blood glucose Only liver, kidney, intestine express G6Pase. Von Gierke disease (G6Pase deficiency): Severe fasting hypoglycaemia + hepatomegaly + lactic acidosis + hyperuricaemia + hyperlipidaemia.
🔴 G6PD deficiency: X-linked; 400 million affected; oxidative haemolysis (primaquine, fava beans, infections) Heinz bodies + bite cells on blood film. Neonatal jaundice. Diagnosis: enzyme assay 2–3 months post-crisis (reticulocytes falsely elevate G6PD during acute haemolysis).
🔴 Classic Galactosaemia (GALT deficiency): Gal-1-P accumulation → E. coli neonatal sepsis + liver failure + cataracts + ovarian failure E. coli sepsis association is pathognomonic. Eliminate galactose/lactose from birth. Ovarian failure occurs in >80% of females despite treatment.
🔴 HFI (Aldolase B deficiency): F1P accumulation → liver damage + hypoglycaemia; patients avoid sweet foods Distinguished from benign fructosuria (fructokinase deficiency — harmless, just fructosuria). Eliminate fructose + sucrose + sorbitol.
🔴 HbA1c ≥6.5% = diabetes diagnosis; reflects 2–3 months average blood glucose Falsely LOW in haemolysis. Falsely HIGH in iron deficiency. Use fructosamine (2–3 week average) when RBC lifespan is abnormal.
🟠 PDH cofactors: “TLC for NAD” — Thiamine-PP (B1), Lipoic acid, CoA (B5), FAD (B2), NAD⁺ (B3) PDH deficiency: lactic acidosis from birth + neurological damage. Treat with ketogenic diet + thiamine. PDH kinase (NADH/Acetyl-CoA activated) inactivates PDH; PDH phosphatase (Ca²⁺/insulin activated) activates it.
🟠 Pyruvate Carboxylase: Biotin cofactor; activated by Acetyl-CoA; first unique gluconeogenic enzyme Mitochondria only. PC deficiency → lactic acidosis + hyperammonaemia. OAA cannot cross mitochondrial membrane → converted to malate or aspartate for export.
🟠 McArdle disease (GSD V): No venous lactate rise on forearm exercise; ammonia DOES rise; second wind Muscle phosphorylase deficiency. Myoglobinuria after exercise → AKI risk. CK very elevated. No hypoglycaemia (muscle cannot supply blood glucose — no G6Pase).
🟠 Pompe disease (GSD II): Acid α-glucosidase (lysosomal); only lysosomal GSD; infant hypertrophic cardiomyopathy Treatment: Alglucosidase alfa (enzyme replacement therapy — effective if started early). Fatal in infancy without treatment.
🟡 Sorbitol pathway: Aldose reductase (NADPH) → sorbitol → sorbitol dehydrogenase (NAD⁺) → fructose Activated in hyperglycaemia in insulin-independent tissues. Cataracts, neuropathy, retinopathy. Target: aldose reductase inhibitors (epalrestat).
🟡 Transketolase requires thiamine (B1/TPP) — functional test for B1 deficiency (erythrocyte transketolase activity) Thiamine deficiency also impairs PDH and α-KG dehydrogenase → Wernicke’s encephalopathy.
🟡 Glucokinase (liver/β-cells): Km 10 mM, not inhibited by G6P, induced by insulin — the glucose sensor MODY 2 (glucokinase mutation) → mild stable hyperglycaemia (set point raised); no progression; no treatment usually.
Mnemonics
“Hexokinase is Humble, Glucokinase is Grand” Humble (Hexokinase): All cells, low Km, inhibited by G6P, constitutive Grand (Glucokinase): Liver/β-cells only, high Km, NOT inhibited by G6P, induced by insulin, only active when glucose is grand (high)
“Please Don’t Forget Glucose” — 4 unique gluconeogenic enzymes P = Pyruvate Carboxylase (Pyruvate→OAA; Biotin; mitochondria) D = Don’t forget PEPCK (OAA→PEP; GTP; cytoplasm) F = Fructose-1,6-Bisphosphatase (F-1,6-BP→F6P; inhibited by AMP + F-2,6-BP) G = Glucose-6-Phosphatase (G6P→Glucose; ER lumen; absent in muscle/brain)
“GALT CATs Get sick Early” — Classic Galactosaemia GALT = enzyme deficiency; C = Cataracts; A = Aminoaciduria (Fanconi); T = Toxic E. coli sepsis; s = Subsequent ovarian failure
“PFK-1’s Fuel Pump” — regulation Pump on: AMP (low energy) + F-2,6-BP (fed/insulin) → ACTIVATE Pump off: ATP (high energy) + Citrate (TCA full) + H⁺ (acidosis) → INHIBIT
Common Mistakes
❌ “All glycolytic enzymes are reversed in gluconeogenesis” ✅ Only 7 of 10 shared. Three irreversible steps need 4 unique bypass enzymes (PC, PEPCK, FBPase-1, G6Pase).
❌ “Muscle glycogen can provide blood glucose during fasting” ✅ Muscle lacks G6Pase → muscle G6P cannot become free glucose → muscle glycogen is local fuel only.
❌ “Benign fructosuria and HFI have the same enzyme deficiency” ✅ Completely different: Benign = Fructokinase (step 1, fructose not phosphorylated, harmless). HFI = Aldolase B (step 2, F1P accumulates, toxic).
❌ “HbA1c is always reliable in all diabetic patients” ✅ Unreliable in haemolysis (falsely low), iron deficiency (falsely high), haemoglobinopathies (assay interference). Use fructosamine or CGM in these situations.
❌ “F-2,6-BP is an intermediate in glycolysis” ✅ F-2,6-BP is purely a regulatory molecule — not a glycolytic or gluconeogenic intermediate. Made and degraded by bifunctional PFK-2/FBPase-2.
5 Practice MCQs
Q1: A patient has jaundice, hepatomegaly, and fasting hypoglycaemia. Liver biopsy shows abnormal amylopectin-like glycogen (few branches). Forearm exercise test — venous lactate rises normally. Which enzyme is deficient?
- A. Glucose-6-Phosphatase (Von Gierke)
- B. Muscle Phosphorylase (McArdle)
- C. Branching Enzyme (Andersen disease)
- D. Debranching Enzyme (Cori disease)
✅ C. Branching Enzyme (Andersen — Type IV GSD)
Amylopectin-like glycogen (abnormal, poorly branched) = branching enzyme deficiency. Forearm test is normal because glycolysis and phosphorylase are intact. Liver failure results from inflammatory response to the foreign-body abnormal glycogen. Type I: Lactate does NOT rise on exercise. Type V: Muscle phosphorylase absent → no lactate from muscle glycogen.
Q2: A 3-day-old neonate has jaundice, hypoglycaemia, and vomiting after breast milk. She develops E. coli septicaemia. Urine Clinitest is positive but glucose oxidase dipstick is negative. Diagnosis and pathogenesis of E. coli sepsis?
- A. HFI; F1P impairs neutrophil function
- B. Classic galactosaemia (GALT deficiency); Gal-1-P impairs neutrophil bactericidal function
- C. Galactokinase deficiency; galactitol impairs neutrophil migration
- D. Pompe disease; lysosomal dysfunction impairs autophagy
✅ B. Classic galactosaemia (GALT deficiency); Gal-1-P impairs neutrophil bactericidal function
Breast milk → lactose → galactose; Clinitest positive (galactose is a reducing sugar) but glucose oxidase negative (galactose ≠ glucose). E. coli neonatal sepsis is pathognomonic for galactosaemia — Gal-1-P impairs neutrophil bactericidal killing. Galactokinase deficiency causes cataracts only (no liver damage, no E. coli association). HFI presents after weaning on fructose/sucrose, not milk.
Q3: Regarding gluconeogenesis regulation — which statement is CORRECT?
- A. Glucagon activates PFK-2, raising F-2,6-BP, which drives gluconeogenesis
- B. Metformin binds PEPCK directly to inhibit its catalytic activity
- C. Acetyl-CoA activates Pyruvate Carboxylase AND simultaneously activates PDH Kinase → inactivates PDH
- D. F-2,6-BP activates Fructose-1,6-Bisphosphatase
✅ C. Acetyl-CoA activates PC AND activates PDH Kinase → inactivates PDH
When Acetyl-CoA is high (fat oxidation in fasting): (1) Activates PC → pyruvate → OAA → gluconeogenesis; (2) Activates PDH Kinase → phosphorylates/inactivates PDH → pyruvate cannot be oxidised. This coordinated switch channels pyruvate into gluconeogenesis. Glucagon INHIBITS PFK-2 (↓ F-2,6-BP). Metformin works via Complex I → ↑ AMP → AMPK → ↓ CRTC2 → ↓ PEPCK/G6Pase transcription. F-2,6-BP INHIBITS FBPase-1.
Q4: A 22-year-old man starts dapsone for leprosy. Three weeks later: Hb 7 g/dL (was 14), jaundice, dark urine, blood film shows Heinz bodies and bite cells. Best timing for confirmatory G6PD assay?
- A. During acute haemolysis — for most accurate result
- B. 2–3 months after recovery — reticulocytes have higher G6PD → false normal during crisis
- C. Immediately — capture abnormal RBC population while active
- D. During next drug challenge (rechallenge test)
✅ B. 2–3 months after recovery
During haemolysis: oldest, most G6PD-deficient RBCs are destroyed selectively. Remaining RBCs are young cells + reticulocytes with higher G6PD activity → false-normal assay. Wait until RBC population returns to normal age distribution (~3 months after the crisis). Rechallenge is inappropriate and potentially harmful. Heinz bodies + bite cells = oxidative haemolysis = G6PD deficiency until proven otherwise.
Q5: A Type 2 diabetic on metformin has HbA1c 8.2%. After treatment of iron deficiency anaemia (Hb normalises, iron stores replete), repeat HbA1c is 6.8% — his glucose diary shows minimal change. Interpretation?
- A. Genuine glycaemic improvement — iron had no effect on HbA1c
- B. Iron deficiency artificially elevated initial HbA1c; true control was ~6.8% throughout; the fall is spurious
- C. Iron supplementation itself lowers blood glucose
- D. Iron deficiency causes falsely LOW HbA1c — his control was worse than 8.2% initially
✅ B. Iron deficiency artificially elevated initial HbA1c
Iron deficiency → ↓ new RBC production → existing RBCs are older → spend more time in circulation → more glycation → falsely HIGH HbA1c. After iron repletion: Burst of new reticulocytes (young RBCs) → less glycation time → HbA1c falls even without glucose change. The glucose diary (not affected by RBC lifespan) confirms no real change. In patients with RBC lifespan disorders, use fructosamine (2–3 week average) or CGM instead of HbA1c.
References
📖 Harper’s Illustrated Biochemistry — Chapters 14–21
📖 Lippincott’s Illustrated Reviews: Biochemistry — Chapters 7–12
📖 Lehninger Principles of Biochemistry — Chapters 13–15
📖 Harrison’s Principles of Internal Medicine — Chapter 397: Diabetes Mellitus
📖 Robbins Basic Pathology — Chapter 7: Genetic and Paediatric Diseases