🗺️ What You Will Learn in This Article
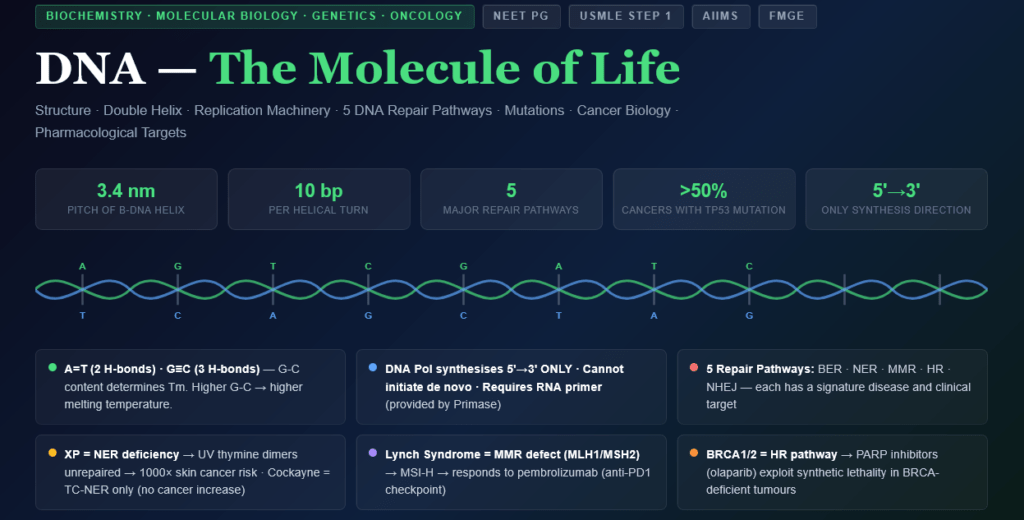
- Describe the complete chemical structure of DNA — bases, nucleotides, backbone, and the double helix
- Explain Watson-Crick base pairing rules and understand why A=T and G≡C
- Trace DNA replication from initiation to termination with every enzyme named and its function
- Distinguish prokaryotic from eukaryotic replication — key enzymes, speed, and regulation
- Explain all five major DNA repair pathways with the diseases caused by each
- Define and classify mutations — point, frameshift, synonymous, missense, nonsense — with clinical examples
- Connect DNA biology to cancer (oncogenes, tumour suppressors, DNA repair defects)
- Explain the molecular basis of pharmacological targets on DNA — antibiotics, chemotherapy drugs
📖 Introduction: Why This Topic Matters in Exams
A 24-year-old woman presents with multiple basal cell carcinomas on her face and arms — unusual for her age. She reports extreme sensitivity to sunlight since childhood and a family history of similar skin cancers. Her skin biopsy shows multiple UV-induced thymidine dimers in tumour DNA. She has xeroderma pigmentosum — a deficiency of the nucleotide excision repair (NER) pathway — and every time her skin is exposed to UV light, the resulting DNA damage cannot be fixed, mutations accumulate, and malignancy follows. This is not just a dermatology case; it is one of the most powerful demonstrations of how DNA repair biochemistry directly determines clinical outcomes.
DNA is the master molecule of life — it carries genetic information, enables inheritance, and when damaged or improperly replicated, drives ageing, cancer, and inherited disease. For medical entrance exams, DNA is not merely theoretical. It underlies the mechanism of multiple antibiotics (fluoroquinolones block DNA gyrase; rifampicin blocks RNA polymerase), chemotherapy agents (alkylating agents, cisplatin, topoisomerase inhibitors), and the entire field of molecular diagnostics (PCR, FISH, Southern blot). Understanding DNA means understanding a huge proportion of pharmacology, pathology, and clinical medicine in one unified framework.
This article covers everything: the double helix structure, base pairing thermodynamics, the complete replication machinery, all five DNA repair pathways, mutation classification, and the clinical and pharmacological significance of each. It is the most comprehensive single reference you will find for DNA in medical education.
🔬 Section 1 — DNA Structure: From Nucleotides to Chromosomes
1.1 The Chemical Building Blocks
The nucleotide is the fundamental unit of DNA. Each nucleotide contains three components:
- Nitrogenous base — carries the genetic information
- Deoxyribose sugar — the 5-carbon sugar (note: 2′-deoxyribose — no OH at C2′)
- Phosphate group — provides the negative charge; forms the backbone
The bases of DNA:
Purines (double ring — pyrimidine + imidazole):
- Adenine (A) — amino group at C6; pairs with Thymine
- Guanine (G) — keto group at C6, amino group at C2; pairs with Cytosine
Pyrimidines (single ring):
- Cytosine (C) — amino group at C4; pairs with Guanine
- Thymine (T) — methyl group at C5; only in DNA (RNA uses Uracil instead)
Memory rule: “CUT the PY — PURe As Gold” — Cytosine, Uracil, Thymine are Pyrimidines; Adenine and Guanine are Purines.
1.2 The Nucleotide Structure and Backbone
Nucleoside = Base + Deoxyribose (no phosphate) Nucleotide = Base + Deoxyribose + Phosphate(s)
- Nucleoside 5′-monophosphate (NMP), diphosphate (NDP), triphosphate (NTP)
- During replication, DNA polymerase incorporates dNTPs (deoxynucleoside triphosphates) — two phosphates are released as pyrophosphate (PPi), which is hydrolysed by pyrophosphatase → drives replication forward (thermodynamic pull)
The phosphodiester backbone:
- Phosphate groups link the 3′-OH of one sugar to the 5′-carbon of the next → 3’→5′ phosphodiester bond
- The backbone is therefore a repeating unit of: 5′-phosphate — sugar — 3′-OH
- DNA strand has directionality: 5′ end (free phosphate) → 3′ end (free OH)
- DNA polymerase synthesises only in the 5’→3′ direction (adds nucleotides to the 3′-OH)
1.3 The Double Helix — Watson and Crick (1953)
Based on:
- X-ray crystallography data from Rosalind Franklin and Maurice Wilkins
- Chargaff’s rules: %A = %T and %G = %C in any double-stranded DNA
- Model building by Watson and Crick
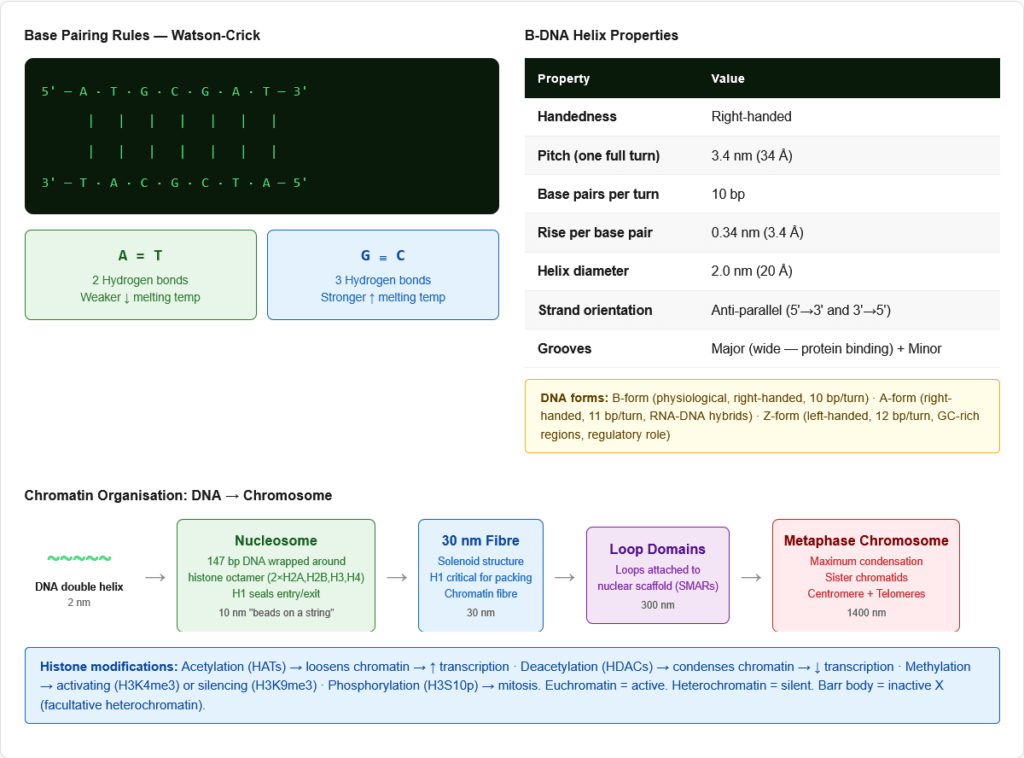
Key structural features of the B-form double helix (the physiologically relevant form):
| Feature | Detail |
|---|---|
| Two anti-parallel strands | One runs 5’→3′, the complementary runs 3’→5′ |
| Right-handed helix | Winds clockwise going away from the observer |
| Pitch (one complete turn) | 3.4 nm (34 Å) |
| Base pairs per turn | 10 bp per helical turn |
| Rise per base pair | 0.34 nm (3.4 Å) |
| Width of helix | 2.0 nm (20 Å) |
| Grooves | Major groove (wide — protein binding sites) and Minor groove (narrow) |
Other DNA conformations:
- A-form: Right-handed; 11 bp/turn; shorter, wider; adopted by RNA-DNA hybrids and dehydrated DNA
- Z-form: Left-handed; 12 bp/turn; adopted in GC-rich regions under high salt; may have regulatory role
1.4 Watson-Crick Base Pairing — The Chemical Basis
Complementary base pairing holds the two strands together via hydrogen bonds:
Adenine — Thymine (A=T): 2 hydrogen bonds
- A amino group (C6) → T carbonyl (C4): H-bond
- A N1 ← T N3-H: H-bond
- Weaker bond — DNA rich in A-T has lower melting temperature (Tm)
Guanine — Cytosine (G≡C): 3 hydrogen bonds
- G N1-H → C N3: H-bond
- G amino (C2) → C carbonyl (C2): H-bond
- G carbonyl (C6) ← C amino (C4): H-bond
- Stronger bond — DNA rich in G-C has higher Tm
- G-C content directly determines thermal stability of DNA
Chargaff’s Rules:
- %A = %T and %G = %C in double-stranded DNA (complementarity rule)
- A+G = T+C (purine = pyrimidine in any dsDNA)
- %G+C content varies between species and is species-specific (and strand-specific)
1.5 DNA Organisation in Eukaryotes: From Helix to Chromosome
Levels of compaction:
- Double helix: 2 nm diameter
- Nucleosome (10 nm “beads on a string”):
- 147 bp of DNA wrapped ~1.65 turns around a histone octamer
- Histone octamer: 2× (H2A + H2B + H3 + H4) — core histones
- H1 (linker histone): Seals the DNA at the entry/exit point; involved in chromatin condensation
- Linker DNA: ~20–80 bp between nucleosomes
- 30 nm fibre (chromatin fibre): Nucleosomes packed into a solenoid structure; H1 histone critical
- 300 nm loop domains: Loops of 30 nm fibre attached to nuclear scaffold (SMAR — scaffold/matrix attachment regions)
- 700 nm fibre: Further condensation
- Metaphase chromosome (1400 nm): Maximum condensation; sister chromatids; 6 μm long
Euchromatin vs Heterochromatin:
- Euchromatin: Lightly staining; actively transcribed; histone acetylation (loosens chromatin → gene expression)
- Heterochromatin: Densely staining; transcriptionally silent; histone methylation (H3K9me3, H3K27me3)
- Constitutive heterochromatin: Permanently silenced (centromeres, telomeres)
- Facultative heterochromatin: Can be activated (Barr body — inactive X chromosome in females)
Histone modifications:
- Acetylation of lysine residues (HATs — histone acetyltransferases): Neutralises positive charge → loosens histone-DNA interaction → ACTIVATES transcription
- Deacetylation (HDACs — histone deacetylases): Restores positive charge → condenses chromatin → SILENCES transcription
- Methylation (HMTs): Can activate (H3K4me3) or silence (H3K9me3) depending on context
- Phosphorylation (H3S10p): Associated with chromosome condensation during mitosis
- Ubiquitination: H2AK119ub1 — gene silencing (Polycomb repression)
Telomeres:
- Repetitive DNA sequences (TTAGGG in humans) at chromosome ends
- Protect chromosomes from degradation and end-to-end fusion
- Telomerase: Reverse transcriptase using an RNA template; maintains telomere length in germ cells, stem cells, and cancer cells
- In somatic cells: Telomerase is suppressed → telomeres shorten with each division → limits the replicative lifespan (Hayflick limit ~50–70 divisions) → contributes to ageing
- Cancer cells: Reactivate telomerase → bypass senescence → immortalisation → one of the hallmarks of cancer
🔬 Section 2 — DNA Replication: Duplicating the Genome
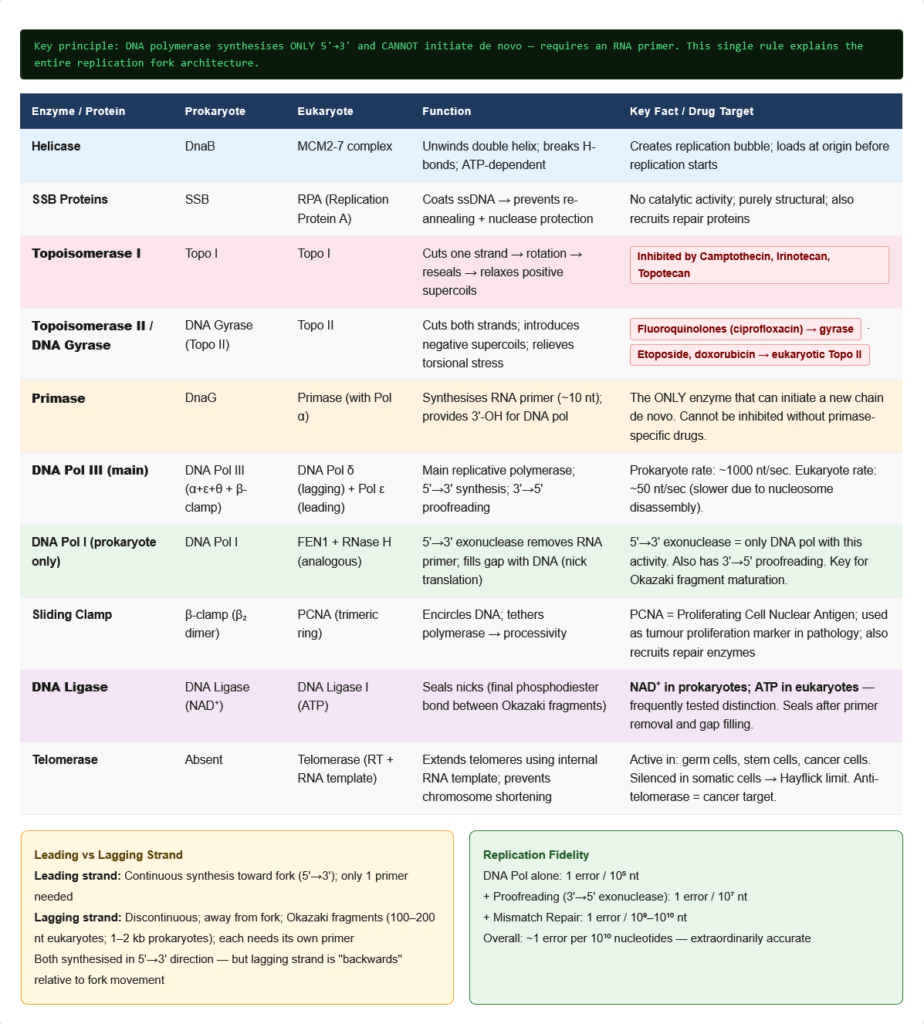
2.1 Fundamental Principles
Semi-conservative replication (Meselson-Stitt experiment, 1958):
- Each parental strand serves as a template for a new complementary strand
- After one round, each daughter DNA contains one parental strand + one new strand
- The key experiment used ¹⁵N / ¹⁴N density labelling in E. coli to prove this
Replication is:
- Semi-conservative (one old strand + one new strand per daughter duplex)
- Bidirectional (proceeds in both directions from each origin)
- Semi-discontinuous (one strand continuous, one strand made in fragments)
- Unidirectional in synthesis (DNA polymerase ALWAYS synthesises 5’→3′)
2.2 The Replication Fork — All Enzymes and Their Roles
Step 1: Origin Recognition and Unwinding
- Origin of replication: Specific DNA sequence where replication begins
- Prokaryotes: Single origin (oriC in E. coli; ~245 bp; AT-rich)
- Eukaryotes: Multiple origins (~tens of thousands in humans) → allows the large genome to replicate in ~8 hours
- Origin Recognition Complex (ORC): In eukaryotes — marks origins; loads MCM helicase
- Helicase (DnaB in prokaryotes; MCM2-7 complex in eukaryotes):
- Unwinds the double helix by breaking hydrogen bonds between base pairs
- Requires ATP hydrolysis
- Creates the replication fork
Step 2: Stabilising the Unwound DNA
- SSB proteins (Single-Strand DNA Binding Proteins):
- Coat the separated single strands to prevent re-annealing and protect from nucleases
- Do not require ATP; function is purely structural
- Topoisomerases:
- Helicase unwinding generates positive supercoiling ahead of the fork
- Topoisomerase I: Cuts one strand, allows rotation, reseals → relaxes supercoils without ATP
- Topoisomerase II (DNA gyrase in bacteria): Cuts BOTH strands, passes another duplex through, reseals → can change linking number by 2 → introduces negative supercoils → relieves positive supercoiling ahead of fork
- Pharmacological targets:
- Fluoroquinolones (ciprofloxacin, levofloxacin): Block DNA gyrase (Topoisomerase II) in bacteria → trapped cleavage complexes → DNA strand breaks → bactericidal
- Camptothecin (irinotecan, topotecan): Block Topoisomerase I in cancer cells
- Etoposide, doxorubicin: Block Topoisomerase II in cancer cells
Step 3: Primer Synthesis
- DNA polymerase CANNOT initiate a new chain — it can only EXTEND an existing 3′-OH
- Therefore, an RNA primer must be synthesised first to provide the 3′-OH
- Primase (DnaG in prokaryotes; primase subunit of primosome in eukaryotes):
- RNA polymerase; synthesises a short RNA primer (~10–12 nt in prokaryotes; ~8–10 nt in eukaryotes)
- Unlike DNA polymerase, primase can initiate a new chain de novo
- The RNA primer provides the essential 3′-OH group for DNA polymerase to extend
Step 4: DNA Synthesis
Prokaryotic DNA Polymerases:
| Polymerase | Primary Role | Special Features |
|---|---|---|
| DNA Pol I | Primer removal and gap filling | Has 5’→3′ exonuclease activity (removes RNA primers); has 3’→5′ proofreading exonuclease |
| DNA Pol II | DNA repair | 3’→5′ exonuclease |
| DNA Pol III | Main replicative polymerase | Core enzyme: α (polymerase) + ε (3’→5′ exonuclease) + θ; β-clamp (processivity); extremely fast (~1000 nt/sec) |
Eukaryotic DNA Polymerases:
| Polymerase | Role |
|---|---|
| DNA Pol α | Primase activity + initiates synthesis; low processivity |
| DNA Pol δ | Main lagging strand polymerase; high fidelity; 3’→5′ exonuclease |
| DNA Pol ε | Main leading strand polymerase; very high fidelity |
| DNA Pol β | Base excision repair (BER) |
| DNA Pol γ | Mitochondrial DNA replication |
| DNA Pol η | Translesion synthesis (bypasses thymine dimers) |
PCNA (Proliferating Cell Nuclear Antigen):
- Eukaryotic equivalent of the β-clamp in prokaryotes
- Homotrimeric ring that encircles DNA and tethers polymerase
- Increases processivity dramatically
- Also recruits repair enzymes, epigenetic factors
- Clinical use: PCNA staining is a marker of cell proliferation in tumour pathology
Step 5: Leading vs Lagging Strand Synthesis
Because the two template strands are anti-parallel but DNA polymerase can only synthesise 5’→3′:
- Leading strand: Synthesised continuously in the 5’→3′ direction toward the fork (same direction as fork movement) — requires only ONE primer
- Lagging strand: Synthesised discontinuously AWAY from the fork in the 5’→3′ direction → produced as Okazaki fragments (1000–2000 nt in prokaryotes; 100–200 nt in eukaryotes), each requiring its own primer
Okazaki fragment processing (prokaryotes):
- DNA Pol III synthesises fragment until it reaches the RNA primer of the preceding fragment
- DNA Pol I arrives → uses its 5’→3′ exonuclease to remove the RNA primer while simultaneously filling in with DNA (nick translation)
- DNA Ligase seals the nick (final phosphodiester bond) — requires NAD⁺ in prokaryotes, ATP in eukaryotes
Eukaryotic Okazaki processing:
- DNA Pol δ synthesises fragment → displaces downstream primer → creates flap
- FEN1 (Flap Endonuclease 1) removes the RNA flap
- DNA Pol δ fills the gap
- DNA Ligase I seals the nick
Step 6: Termination
Prokaryotes:
- Termination sequences (ter sites in E. coli) with specific terminator proteins (Tus) halt the replication fork
- The two forks from the single origin meet at the terminus and are resolved by Topoisomerase II
Eukaryotes:
- No specific termination sequences — forks from adjacent origins simply meet
- End-replication problem: The lagging strand template cannot be fully replicated at the 5′ end (primase cannot start without a template overhang) → progressive shortening of chromosome ends → TELOMERE PROBLEM
- Telomerase solves this in germ cells: extends the 3′-overhang using an internal RNA template → provides the template for lagging strand completion
2.3 Fidelity of DNA Replication
Error rate of replication machinery:
- DNA polymerase alone: ~1 error per 10⁵ nt (10⁻⁵)
- After proofreading (3’→5′ exonuclease): ~1 error per 10⁷ nt (10⁻⁷)
- After mismatch repair (MMR): ~1 error per 10⁹–10¹⁰ nt (10⁻⁹ to 10⁻¹⁰)
- Total fidelity: ~1 error per 10¹⁰ nt synthesised
2.4 Speed and Scale of Replication
- E. coli: One origin; ~4.6 Mbp; replication rate ~1000 nt/sec; completes in ~40 min
- Human genome: ~6,400 Mbp (diploid); ~30,000+ origins; rate ~50 nt/sec (slower due to nucleosome disassembly/reassembly); completes in ~8 hours (S phase)
⚙️ Section 3 — DNA Damage: Types and Causes
Before repair can occur, understand what types of damage arise:
Types of DNA Damage
| Damage Type | Cause | Structure | If Unrepaired |
|---|---|---|---|
| Thymine dimers (CPDs) | UV radiation (UVB 280–315 nm) | Two adjacent thymines covalently linked (cyclobutane ring) | Blocks replication; causes mutations → skin cancer |
| 6-4 photoproducts | UV radiation | Another UV-induced lesion between adjacent pyrimidines | Similar to thymine dimers |
| Alkylation | Alkylating agents (cancer drugs, carcinogens) | O⁶-methylguanine (most mutagenic); N7-methylguanine | Mispairing (O⁶-meG pairs with T) → G→A transitions |
| Deamination | Spontaneous; nitrous acid | Cytosine → Uracil; 5-methylcytosine → Thymine | C→U transition; CpG → TpG (most common human mutation) |
| Depurination | Spontaneous hydrolysis | Loss of purine base → apurinic/apyrimidinic (AP) site | Blocks replication; frame shift or transversion mutations |
| Oxidative damage | ROS (mitochondria, ionising radiation) | 8-oxoguanine (8-oxo-G) — mispairs with A → G:C → T:A transversion | Mutations |
| Single-strand break | Ionising radiation, oxidative damage, topoisomerase | Break in phosphodiester backbone | Block replication if unrepaired |
| Double-strand break (DSB) | Ionising radiation, chemotherapy, replication errors | Both strands cut | Most dangerous; chromosomal rearrangements, cell death |
| Interstrand crosslink | Cisplatin, nitrogen mustards, mitomycin C | Covalent links between the two strands | Blocks both replication and transcription |
| Intrastrand adducts | Cisplatin | d(GpG) or d(ApG) cisplatin adducts | Distorts helix; blocks polymerase |
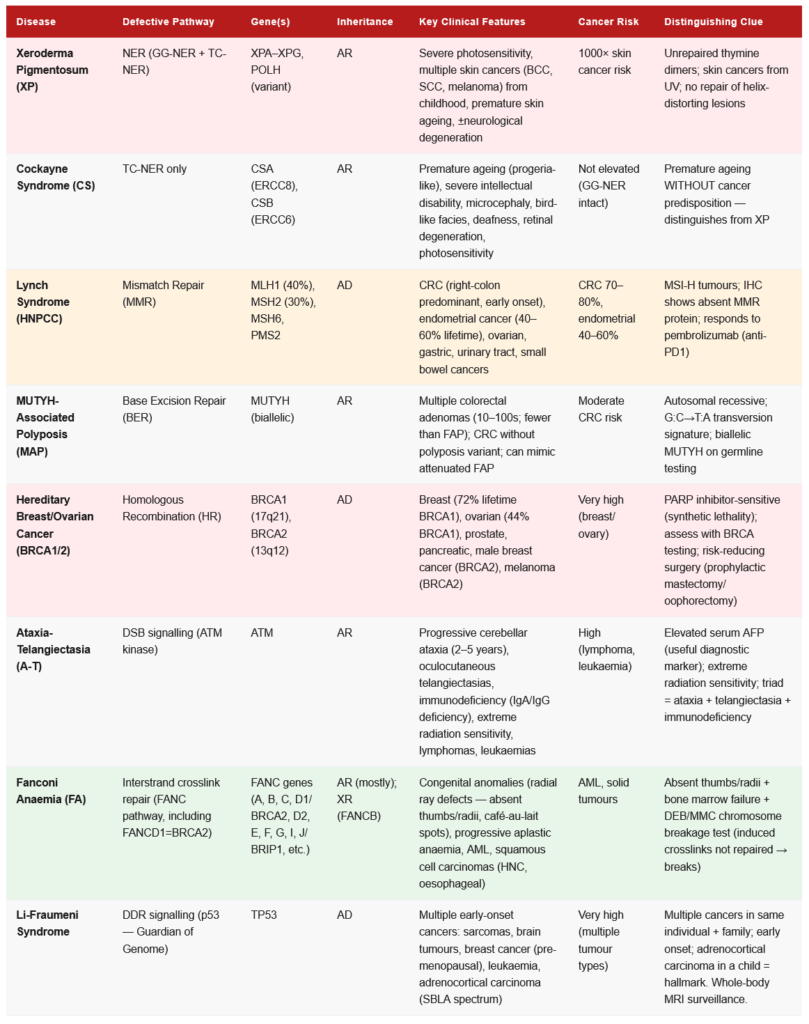
⚙️ Section 4 — DNA Repair Pathways: The Five Essential Systems

Repair Pathway 1: Base Excision Repair (BER)
Substrates: Small, non-helix-distorting lesions — deaminated bases (uracil in DNA), oxidised bases (8-oxoG), alkylated bases (3-methyladenine), depurination/depyrimidination (AP sites)
Pathway:
- DNA Glycosylase — recognises and removes the damaged base (cuts N-glycosidic bond) → creates AP site (abasic site). Different glycosylases for different lesions:
- UNG (Uracil DNA Glycosylase): removes Uracil (from C deamination)
- OGG1: removes 8-oxoguanine
- AAG: removes 3-methyladenine
- AP Endonuclease (APE1) — cleaves phosphodiester backbone 5′ to AP site → creates a strand nick
- DNA Polymerase β — fills in the single nucleotide gap (short-patch BER) or 2–8 nucleotides (long-patch BER)
- DNA Ligase III / XRCC1 — seals the nick
- FEN1 (long-patch BER) — removes the displaced flap
Clinical relevance:
- BER is the primary defence against oxidative DNA damage (a continuous threat from normal metabolism)
- MUTYH (MutY Homolog): Removes Adenine opposite 8-oxoG before repair. MUTYH-associated polyposis (MAP): Biallelic MUTYH mutations → failure to correct 8-oxoG lesions → excess G→T transversions → colorectal cancer syndrome (autosomal recessive; multiple colorectal adenomas/carcinomas; must be distinguished from FAP)
- PARP (Poly(ADP-ribose) Polymerase): Detects single-strand breaks → recruits BER machinery. PARP inhibitors (olaparib, rucaparib, niraparib): Used in BRCA1/2-deficient cancers (ovarian, breast, prostate, pancreatic) — the principle of synthetic lethality: BRCA-deficient cells cannot repair DSBs by HR; when PARP is also inhibited, SSBs accumulate and collapse replication forks → DSBs → cell death selectively in BRCA-deficient tumours.
Repair Pathway 2: Nucleotide Excision Repair (NER)
Substrates: Large, helix-distorting lesions — thymine dimers (CPDs, 6-4PP), bulky chemical adducts (benzo[a]pyrene-dG adducts from cigarette smoke), cisplatin adducts
Two sub-pathways:
- Global Genome NER (GG-NER): Scans the entire genome for distortions; uses XPC-RAD23B as damage sensor
- Transcription-Coupled NER (TC-NER): Repairs lesions in the template strand of actively transcribed genes; uses RNA polymerase stalling at the lesion as the damage signal; faster — explains why the template strand of active genes is repaired preferentially
Pathway (common steps after damage recognition):
- Damage recognition:
- GG-NER: XPC-RAD23B detects helix distortion
- TC-NER: RNA Pol II stalls at lesion → CSA and CSB (Cockayne syndrome proteins) are recruited
- TFIIH complex recruited (contains XPB and XPD helicases) → unwinds DNA ~30 bp around the lesion
- XPA and RPA — verify the lesion and stabilise the bubble
- XPG (3′ endonuclease) and XPF-ERCC1 (5′ endonuclease) — make dual incisions:
- XPF-ERCC1 cuts ~25 nt 5′ of the lesion
- XPG cuts ~8 nt 3′ of the lesion
- An oligonucleotide of ~25–30 nt containing the lesion is excised
- DNA Polymerases δ/ε + PCNA/RPA fill the gap
- DNA Ligase I (or Ligase III/XRCC1) seals the nick
Clinical diseases of NER deficiency:
Xeroderma Pigmentosum (XP) — Autosomal Recessive:
- Mutations in XPA–XPG genes
- Cannot repair UV-induced thymine dimers → progressive accumulation of mutations → skin cancers (basal cell carcinoma, squamous cell carcinoma, melanoma) on sun-exposed areas
- 1000× increased risk of skin cancer; onset in childhood
- Features: Acute sunburn reactions, photophobia, progressive pigmentary changes, xerosis (dry skin), premature ageing of skin
- Some variants (XPD, XPB mutations) also affect transcription (TFIIH is needed for transcription) → neurological degeneration (XP neurological form)
- Treatment: Lifetime sun avoidance (protective clothing, UV-blocking windows), regular skin surveillance, surgical excision of tumours
Cockayne Syndrome (CS) — Autosomal Recessive:
- Mutations in CSA (ERCC8) or CSB (ERCC6) genes — TC-NER specific
- Global genome NER is intact → UV-induced skin cancers are NOT elevated
- Preferential failure to repair transcribed genes → premature ageing, severe intellectual disability, microcephaly, photosensitivity without cancer predisposition, progressive neuropathy
- Features: Cachectic dwarfism, “bird-like facies,” senile appearance from childhood, deafness, retinal degeneration, cataracts
- No skin cancer risk (because GG-NER is intact — the cancer risk in XP is from failure to repair the whole genome)
Trichothiodystrophy (TTD):
- XPB or XPD mutations — but these proteins also function in TFIIH for transcription
- Brittle hair and nails (sulfur deficiency), photosensitivity, ichthyosis, intellectual disability, short stature
- Sulfur-deficient hair gives “tiger tail” appearance under polarised microscopy
Repair Pathway 3: Mismatch Repair (MMR)
Substrates: Replication errors — mismatched base pairs (G-T, A-C mismatches after replication errors), insertion/deletion loops (IDLs) in microsatellite regions
Key challenge: MMR must distinguish the newly synthesised (error-containing) strand from the parental (template) strand to know which base to remove.
Prokaryotic MMR (model for understanding eukaryotic MMR):
- Dam methylase methylates adenine at GATC sequences — methylation is not immediately present on newly synthesised strand (lag) → newly synthesised strand is unmethylated → MutH recognises unmethylated strand = new strand → repairs on the new strand
- MutS: Recognises mismatches; MutS forms complex at mismatch
- MutL: Mediator; recruits MutH
- MutH: Endonuclease; cleaves the unmethylated (new) strand at hemimethylated GATC site
- Exonuclease removes strand from nick to past the mismatch
- DNA Pol III + SSB refills
- DNA Ligase seals
Eukaryotic MMR:
- No Dam methylase — strand discrimination uses nicks in the newly synthesised strand (from Okazaki fragment processing and PCNA)
- MSH2-MSH6 (MutSα): Recognises base-base mismatches and small IDLs
- MSH2-MSH3 (MutSβ): Recognises larger IDLs
- MLH1-PMS2 (MutLα): Endonuclease; cleaves new strand
- EXO1: Exonuclease; removes the mismatch-containing strand
- DNA Pol δ + PCNA: Fills the gap
- DNA Ligase I: Seals
Clinical consequences of MMR deficiency — Lynch Syndrome:
Lynch Syndrome (Hereditary Non-Polyposis Colorectal Cancer — HNPCC):
- Autosomal dominant — most common hereditary colorectal cancer syndrome (2–5% of all CRC)
- Caused by germline mutations in MMR genes: MLH1 (most common, ~40%), MSH2 (~30%), MSH6, PMS2
- Mechanism: One defective MMR allele inherited (germline) + second hit (somatic) in the other allele → complete MMR loss → microsatellite instability (MSI-H) — replication errors in microsatellite repeats accumulate → frameshift mutations in tumour suppressor genes → cancer
- Clinical features: CRC (right-colon predominant), endometrial cancer (most common extracolonic; lifetime risk ~40–60%), ovarian, gastric, urinary tract, small bowel, biliary tract, brain (Turcot variant with CNS tumours) cancers
- Amsterdam II criteria (3-2-1 rule): ≥3 relatives with Lynch syndrome-associated cancers; ≥2 successive generations affected; ≥1 case diagnosed before age 50; FAP excluded
- Bethesda guidelines: Help identify tumours for MSI testing
- Diagnosis: Tumour MSI testing (PCR of microsatellite markers) → MSI-H positive + IHC for MMR proteins → germline testing
- Surveillance: Colonoscopy every 1–2 years from age 20–25; annual endometrial sampling from age 30–35; annual urinalysis
Microsatellite Instability (MSI):
- MSI-High (MSI-H): Loss of MMR function → high mutation burden → increased neoantigens → IMMUNE RECOGNITION
- MSI-H tumours respond dramatically to immune checkpoint inhibitors (pembrolizumab) — FDA approved for any MSI-H/dMMR solid tumour (tumour-agnostic approval)
- MSI-H is also associated with better prognosis in stage II CRC (paradoxically — because of immune surveillance)
Constitutional MMR Deficiency (CMMRD):
- Biallelic (homozygous) germline MMR mutations → early onset of cancers in childhood (brain tumours, lymphomas, colorectal cancers); café-au-lait spots; NF1 phenotype overlap
Repair Pathway 4: Homologous Recombination (HR)
Substrates: Double-strand breaks (DSBs) — the most dangerous form of DNA damage When it operates: S phase and G2 (requires sister chromatid as template for high-fidelity repair) Fidelity: Error-free (uses sister chromatid as template)
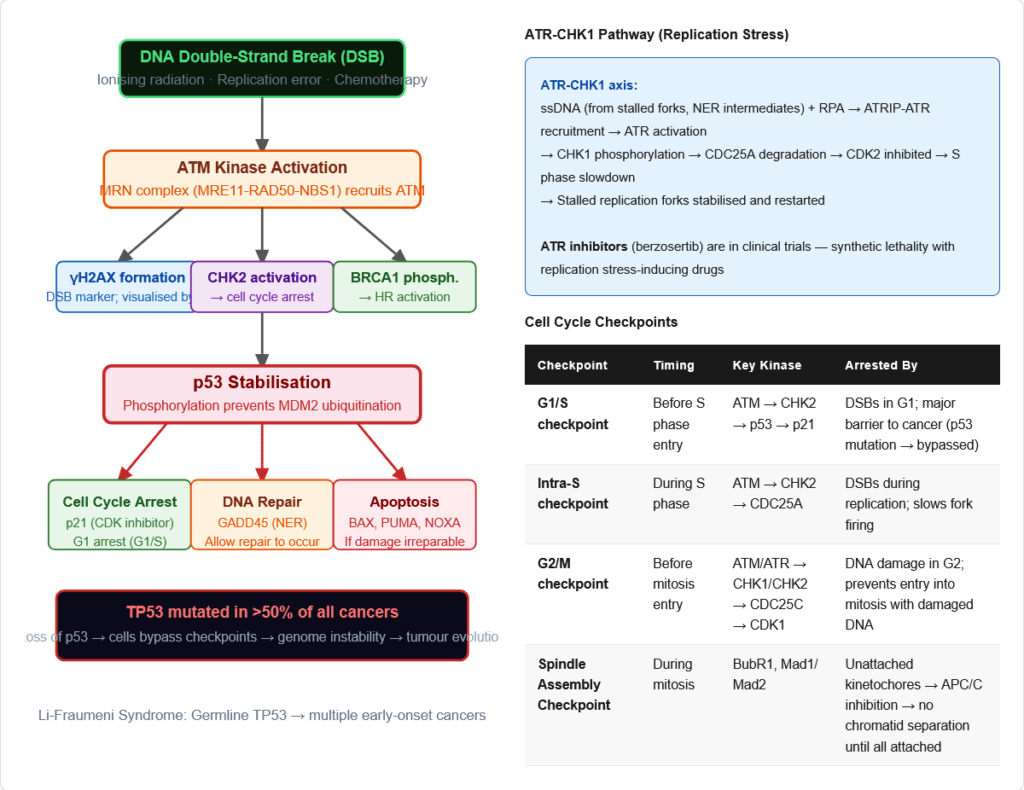
Pathway:
- DSB recognition: MRN complex (MRE11-RAD50-NBS1) detects DSB; recruits ATM kinase
- ATM activation: ATM phosphorylates H2AX (→ γH2AX — a marker of DSBs); activates CHK2 → p53 → cell cycle arrest (G2/M checkpoint)
- End resection: MRN + CTIP generate 3′ single-strand overhangs; RPA coats the ssDNA
- RAD51 loading: BRCA2 (FANCD1) loads RAD51 onto ssDNA, displacing RPA → RAD51 filament = key step
- Strand invasion: RAD51 filament invades homologous duplex (sister chromatid) → D-loop intermediate
- DNA synthesis and resolution: New DNA is synthesised using the homologous template; Holiday junction structures are resolved by resolvases (GEN1, MUS81)
- Result: Error-free repair — the sequence is restored exactly
Key proteins:
- BRCA1: Scaffold/mediator; promotes end resection; helps recruit PALB2-BRCA2; also involved in NHEJ regulation; acts as tumour suppressor
- BRCA2: Directly loads RAD51 onto ssDNA — essential for HR; contains 8 BRC repeats that bind RAD51
- PALB2: Partner and localiser of BRCA2 — connects BRCA1 to BRCA2; loss → BRCA2 not recruited to DSBs
Clinical Importance — BRCA1/2 Mutations:
- BRCA1 (chromosome 17q21): Hereditary breast/ovarian cancer syndrome; lifetime breast cancer risk ~72%; ovarian cancer risk ~44%; also prostate, pancreatic
- BRCA2 (chromosome 13q12): Similar spectrum; also male breast cancer; pancreatic; prostate (Gleason >7); melanoma
- Fanconi Anaemia (FA): Biallelic mutations in FANC genes (including FANCD1 = BRCA2) → congenital anomalies (radial ray defects — absent thumbs), progressive aplastic anaemia, cancer predisposition (AML, squamous cell carcinomas)
- PARP inhibitors: Exploit synthetic lethality in BRCA-deficient tumours (see BER section)
- Nijmegen Breakage Syndrome (NBS): NBN (NBS1) mutations → microcephaly, immunodeficiency, radiation sensitivity, lymphoma predisposition
- Ataxia-Telangiectasia (A-T): ATM mutations → cerebellar ataxia (age 2–5), telangiectasias, immunodeficiency, extreme radiation sensitivity, lymphomas, leukaemias, elevated AFP, serum AFP useful diagnostic marker
Repair Pathway 5: Non-Homologous End Joining (NHEJ)
Substrates: Double-strand breaks When it operates: All cell cycle phases (G1, S, G2) — the primary DSB repair pathway in G1 Fidelity: Error-prone — can introduce small insertions/deletions at the join site
Pathway:
- DSB recognition: Ku70/Ku80 heterodimer binds the free DNA ends — rapid (within seconds); recruits DNA-PKcs
- DNA-PKcs (DNA-dependent Protein Kinase, catalytic subunit): Large kinase; phosphorylates itself and other proteins; holds the DNA ends in proximity
- End processing: Artemis nuclease (with DNA-PKcs) processes incompatible ends (hairpins, overhangs) → trimming
- Polymerase fill-in: Pol μ or Pol λ fills any gaps
- Ligation: XRCC4-DNA Ligase IV complex (with XLF/Cernunnos) ligates the ends
- Result: DSBs are rejoined, but often with loss of 1–10 nt at the junction
Physiological uses of NHEJ:
- V(D)J recombination (antigen receptor diversity in B and T lymphocytes) — RAG1/RAG2 create programmed DSBs → NHEJ rejoins the segments → creates diverse antibody/TCR repertoire
- Class switch recombination in B cells (IgM → IgG, IgA, IgE)
Clinical relevance:
- Severe Combined Immunodeficiency (SCID): Mutations in Artemis, DNA-PKcs, LIG4, XLF, XRCC4 → failed V(D)J recombination → no functional B or T cells
- Radiation therapy exploits NHEJ — ionising radiation creates DSBs → tumour cells cannot repair → cell death; rapidly dividing cells (tumours) are more sensitive
🔬 Section 5 — Mutations: Types, Consequences, and Clinical Significance
5.1 Classification of Mutations
By Scale:
- Point mutation: Single nucleotide change
- Insertion or deletion (indel): Addition or removal of one or more nucleotides
- Inversion: Segment reversed
- Translocation: Segment moved to different chromosome
- Amplification: Increased copy number of a gene region
Point Mutations by Chemistry:
- Transition: Purine replaced by purine, or pyrimidine by pyrimidine (A↔G; C↔T) — more common
- Transversion: Purine replaced by pyrimidine, or vice versa (A/G ↔ C/T) — less common
Point Mutations by Functional Consequence:
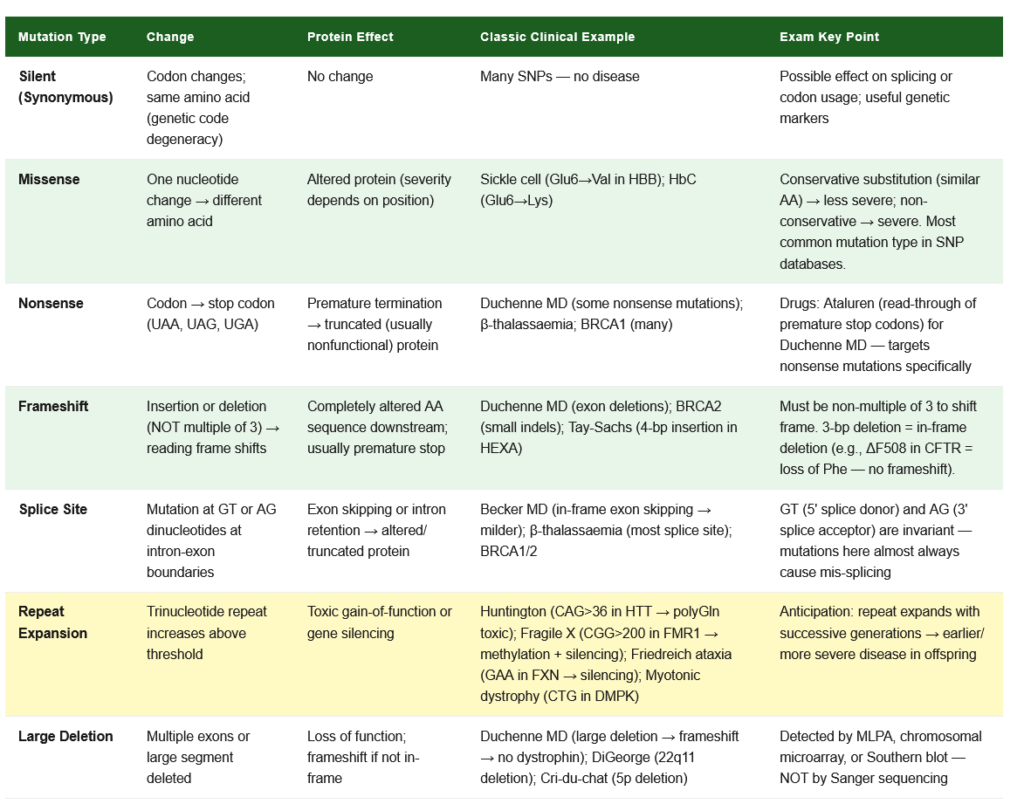
| Type | Definition | Protein Effect | Clinical Example |
|---|---|---|---|
| Silent (synonymous) | Codon changes but same amino acid coded (degeneracy of genetic code) | No change in protein | Often no disease effect; useful genetic markers |
| Missense | Codon change → different amino acid | Altered protein (function depends on position) | Sickle cell — Glu→Val (HbS); Haemoglobin C — Glu→Lys |
| Nonsense | Codon → stop codon (UAA, UAG, UGA) | Premature termination → truncated protein | Duchenne MD, β-thalassaemia, BRCA1 (many mutations) |
| Frameshift | Insertion or deletion (not multiple of 3) → shift in reading frame | Completely altered protein downstream; often creates premature stop → nonfunctional | Duchenne MD (exon deletions), BRCA2 (small indels) |
| Splice site | Mutation at intron-exon boundary | Altered splicing → exon skipping or intron retention | Becker MD (exon skipping), many thalassaemias, BRCA1/2 |
5.2 Consequences of Mutations
Effect on gene product:
- Loss-of-function (LOF): Protein activity is reduced or absent; usually recessive (one copy usually sufficient); tumour suppressors follow this pattern
- Gain-of-function (GOF): Protein gains new or increased activity; usually dominant (one mutant allele sufficient); oncogenes follow this pattern; also Huntington disease (expanded CAG → toxic polyglutamine)
Key clinical examples:
| Mutation | Gene | Type | Disease |
|---|---|---|---|
| Glu6Val | HBB (β-globin) | Missense | Sickle cell anaemia (HbS) |
| Glu6Lys | HBB | Missense | Haemoglobin C disease |
| CAG repeat expansion | HTT | Expansion | Huntington disease (>36 repeats) |
| CTG repeat expansion | DMPK | Expansion | Myotonic dystrophy type 1 |
| GAA repeat expansion | FXN | Expansion | Friedreich’s ataxia |
| CGG repeat expansion | FMR1 | Expansion | Fragile X syndrome (>200 repeats) |
| Del Phe508 (ΔF508) | CFTR | Deletion (3 nt) | Cystic fibrosis (most common CF mutation) |
| Codon 1309 | APC | Frameshift | Familial adenomatous polyposis (FAP) |
5.3 Carcinogens and Mutation Signatures
Understanding how carcinogens cause specific mutations:
| Carcinogen | Type of Damage | Mutation | Cancer |
|---|---|---|---|
| UV radiation (UVB) | Thymine dimers → C→T transitions at TT | C→T at pyrimidine dimers | Skin cancers (BCC, SCC, melanoma) |
| Tobacco smoke (benzo[a]pyrene) | G adducts → G→T transversions | G:C → T:A transversions | Lung cancer (TP53 mutations at CpG sites) |
| Aflatoxin B1 | G adducts → G→T transversions at codon 249 | TP53 R249S mutation | Hepatocellular carcinoma (endemic in sub-Saharan Africa, China) |
| Alkylating agents | O⁶-methylguanine → pairs with T | G:C → A:T transitions | |
| Spontaneous deamination | CpG sites — 5-methylcytosine → Thymine | C→T at CpG | Most common mutation in human disease (~1/3 of all point mutations) |
5.4 The COSMIC Mutational Signatures
The Catalogue of Somatic Mutations in Cancer (COSMIC) identifies >60 mutational signatures in cancer genomes, each reflecting a specific mutational process. Clinically relevant examples:
- Signature 1: Spontaneous deamination of 5-methylcytosine (C→T at CpG) — clock-like, accumulates with age
- Signature 4: Tobacco carcinogen adducts — G→T transversions; dominant in lung and head/neck cancers
- Signature 7: UV radiation — C→T and CC→TT at pyrimidine doublets; dominant in skin cancers
- Signature 6, 15, 20, 21, 26: MMR deficiency — associated with MSI-H
🏥 Section 6 — DNA and Disease: Oncology Connections
DNA Damage Response (DDR) and Cancer
The DDR is a signalling network triggered by DNA damage that activates repair, cell cycle checkpoints, and apoptosis if damage is irreparable:
ATM-CHK2-p53 pathway (primarily activated by DSBs):
- DSB → ATM activation (phosphorylates H2AX, CHK2, BRCA1)
- CHK2 → phosphorylates CDC25A/C (degraded) → CDK2/CDK1 inhibited → S/G2 checkpoint
- ATM → p53 phosphorylation (stabilises p53 by preventing MDM2 binding)
- p53 → induces p21 (CDK inhibitor → G1 arrest) + GADD45 (NER) + BAX/PUMA (apoptosis)
ATR-CHK1 pathway (primarily activated by ssDNA — replication stress, NER intermediates):
- ssDNA-RPA → ATRIP-ATR recruitment → ATR activation
- ATR → CHK1 phosphorylation → CDC25A degradation → S phase slowdown
- Replication fork stabilisation and restart
p53 — “Guardian of the Genome”:
- Tumour suppressor — mutated in >50% of all cancers (most frequently mutated gene in cancer)
- Normal function: Transcription factor that activates DNA repair genes, cell cycle arrest genes (p21), and apoptosis genes (BAX, PUMA, NOXA)
- In cancer: LOF mutations → cells cannot respond to DNA damage → genome instability → additional mutations → tumour progression
- Li-Fraumeni Syndrome: Germline TP53 mutations → multiple early-onset cancers (sarcomas, brain tumours, breast cancer, leukaemia, adrenocortical carcinoma)
Hallmarks of Cancer and DNA
Multiple hallmarks of cancer connect directly to DNA biology:
- Genomic instability — from defective repair pathways
- Replicative immortality — from telomerase reactivation
- Tumour suppressor inactivation — from mutations in TP53, RB1, APC, BRCA1/2, PTEN, etc.
- Oncogene activation — from mutations in KRAS, BRAF, EGFR, MYC, etc.
- Microsatellite instability — from MMR deficiency → MSI-H tumours
🔬 Section 7 — Pharmacological Targets on DNA
Antibiotics Targeting DNA Metabolism
| Drug | Target | Mechanism | Clinical Use |
|---|---|---|---|
| Fluoroquinolones (Ciprofloxacin, Levofloxacin, Moxifloxacin) | Bacterial DNA gyrase (Topoisomerase II) + Topoisomerase IV | Stabilise cleavage complex → DSBs → bactericidal | Gram-negative UTIs, respiratory, GI infections; ciprofloxacin for anthrax |
| Rifampicin | Bacterial RNA polymerase β-subunit | Blocks initiation of transcription (NOT directly DNA) | TB, meningococcal prophylaxis; NOT for ongoing transcription |
| Metronidazole | DNA (indirect) | Reduced to reactive nitro radicals in anaerobes → strand breaks | Anaerobic infections, H. pylori, C. difficile, Giardia, Trichomonas |
Chemotherapy Drugs Targeting DNA
| Drug Class | Drug | Mechanism | Tumour Types |
|---|---|---|---|
| Alkylating agents | Cyclophosphamide, Chlorambucil, Melphalan | Alkylate N7-G or O⁶-G → interstrand crosslinks → blocked replication → apoptosis | Lymphomas, leukaemias, ovarian, breast |
| Platinum compounds | Cisplatin, Carboplatin, Oxaliplatin | Intrastrand d(GpG) crosslinks → helix distortion → NER substrate → if NER deficient → apoptosis | Testicular (cisplatin — curative), ovarian, lung, head/neck, colorectal (oxaliplatin) |
| Topoisomerase I inhibitors | Irinotecan, Topotecan | Stabilise Topo I cleavage complex → persistent SSBs → replication fork collapse → DSBs → apoptosis | Colorectal (irinotecan), ovarian/lung (topotecan) |
| Topoisomerase II inhibitors | Etoposide, Doxorubicin | Stabilise Topo II cleavage complex → DSBs | Testicular (etoposide), lymphomas, breast, sarcomas (doxorubicin) |
| Antimetabolites | 5-FU, Methotrexate, Gemcitabine | Inhibit nucleotide synthesis (TS, DHFR, RNR) → deplete dNTP pool → replication failure | Colorectal (5-FU), many cancers (MTX), pancreatic (gemcitabine) |
| PARP inhibitors | Olaparib, Rucaparib, Niraparib | Inhibit PARP → SSBs not repaired → replication fork collapse → DSBs → synthetic lethality in BRCA-deficient cells | BRCA1/2-deficient breast, ovarian, prostate, pancreatic |
| Alkylating (nitrosoureas) | Temozolomide, Carmustine | Alkylate O⁶-G → G:T mispairing → MMR futile cycles → DSBs | Glioblastoma (temozolomide), brain tumours (carmustine) |
🔄 Section 8 — Connections to Other Topics
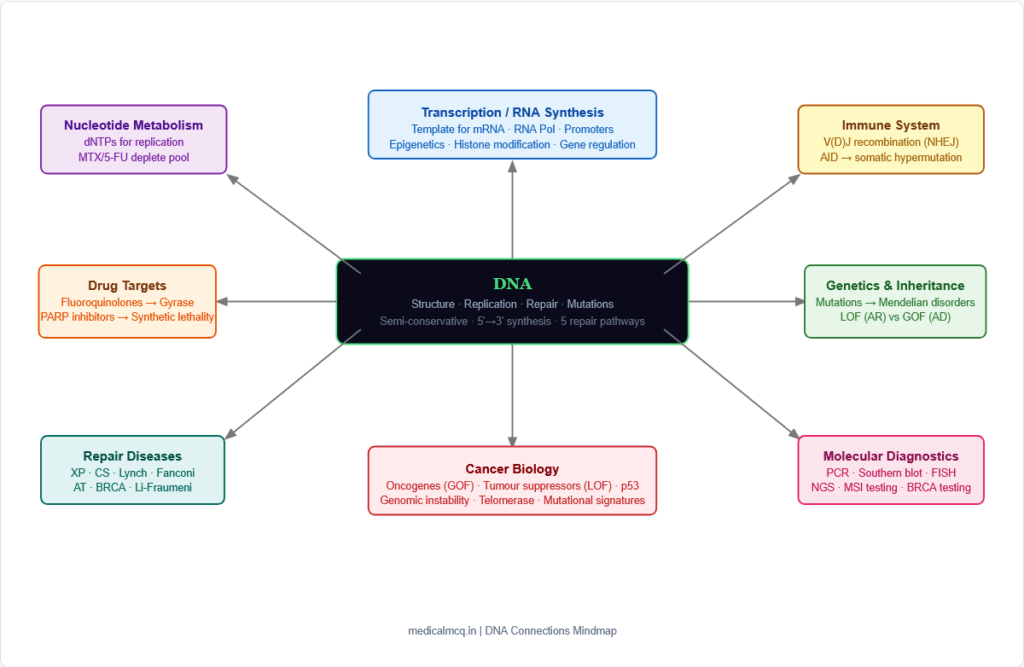
→ Gene Expression / Transcription — DNA is the template for RNA synthesis. The promoter sequences, transcription factor binding sites, and enhancers are all features of the DNA double helix. Epigenetic modifications (methylation, histone modification) that regulate transcription are all applied to DNA. Understanding DNA structure is the foundation for understanding gene regulation.
→ Molecular Diagnostics — PCR uses DNA polymerase in vitro; Southern blotting detects specific DNA sequences; FISH uses DNA hybridisation; gene sequencing (Sanger, NGS) depends on DNA template. All of these diagnostic tools flow directly from understanding DNA replication and structure.
→ Chromosomal Disorders — The packaging of DNA into chromosomes and the regulation of chromosome segregation are DNA problems. Aneuploidy (Down syndrome, Turner, Klinefelter), chromosomal translocations (Philadelphia chromosome in CML — BCR-ABL), and chromosomal deletions (cri-du-chat, DiGeorge) all involve structural alterations of DNA organisation.
→ Mendelian Genetics — Mutations in DNA are the molecular basis of all Mendelian disorders. Autosomal dominant (one allele sufficient — usually GOF), autosomal recessive (both alleles must be mutated — usually LOF), X-linked (gene on X chromosome — males have one copy), and mitochondrial inheritance (mitochondrial DNA — maternal) all depend on understanding DNA mutations.
→ Nucleotide Metabolism — The dNTPs incorporated during DNA replication come from nucleotide synthesis pathways. Drugs that inhibit purine or pyrimidine synthesis (methotrexate, 5-FU, hydroxyurea) act by depleting the dNTP pool needed for replication. Ribonucleotide reductase converts NDPs to dNDPs (the key dNTP biosynthetic step).
→ Immunology — V(D)J recombination (uses NHEJ to generate antibody diversity), somatic hypermutation (AID deaminase creates mutations in antibody variable regions → affinity maturation), and class switch recombination (NHEJ-based) are all DNA repair processes in the service of the immune system.
🎯 High-Yield Exam Facts
🔴 DNA polymerase synthesises ONLY in the 5’→3′ direction and requires a primer (3′-OH) to extend This single rule explains the entire replication fork mechanism — why a primer is needed, why the lagging strand is discontinuous, why Okazaki fragments exist, and why the end-replication problem occurs.
🔴 Semi-conservative replication proven by Meselson-Stitt experiment (1958) using ¹⁵N/¹⁴N CsCl density gradient Each daughter cell receives one parental strand and one new strand. This is the foundational experiment proving how DNA is replicated.
🔴 G-C base pairs have 3 hydrogen bonds; A-T pairs have 2. Higher G-C content = higher melting temperature (Tm) The Tm of DNA is directly calculated from G-C content: Tm = 69.3 + 0.41 × (%G+C). Used in PCR primer design.
🔴 Xeroderma Pigmentosum = NER deficiency → cannot repair thymine dimers → UV-induced skin cancers The XP genes (XPA–XPG) are direct exam targets. The condition demonstrates that NER is the primary defence against UV damage. 1000× increased skin cancer risk.
🔴 Lynch Syndrome = MMR deficiency (MLH1, MSH2, MSH6, PMS2 mutations) → MSI-H → colorectal + endometrial cancers Most common hereditary CRC syndrome. MMR-deficient (dMMR) / MSI-H tumours respond to pembrolizumab (anti-PD1) — a direct clinical application of DNA repair biochemistry.
🔴 BRCA1/2 = HR pathway → PARP inhibitors exploit synthetic lethality in BRCA-deficient tumours BRCA-deficient cells cannot perform HR. When PARP is inhibited, SSBs → DSBs → cell death selectively in BRCA-deficient tumours. Olaparib, rucaparib, niraparib are FDA-approved PARP inhibitors.
🔴 Fluoroquinolones inhibit bacterial DNA gyrase (Topoisomerase II) — prokaryote-specific target Human cells use Topoisomerase II but fluoroquinolones have much greater affinity for the bacterial enzyme (selective toxicity). The mechanism — stabilising cleavage complexes — causes DSBs → bactericidal.
🟠 Ataxia-Telangiectasia = ATM kinase deficiency → cerebellar ataxia + telangiectasias + radiation hypersensitivity + lymphoma + elevated AFP ATM is the master kinase activated by DSBs. Its loss means cells cannot respond to ionising radiation or other DSB-causing agents → clinical radiosensitivity + immune defects + cancer predisposition.
🟠 The lagging strand is synthesised as Okazaki fragments (1–2 kb in prokaryotes; 100–200 nt in eukaryotes) Each fragment requires its own RNA primer. Primer removal (by DNA Pol I’s 5’→3′ exonuclease in prokaryotes; by FEN1 in eukaryotes) and gap filling creates the continuous lagging strand.
🟠 DNA Ligase requires NAD⁺ in prokaryotes and ATP in eukaryotes A frequently tested distinction. In both cases, the energy is used to form the last phosphodiester bond, sealing the nick between Okazaki fragments (or at the end of repair patch synthesis).
🟠 PCNA (Proliferating Cell Nuclear Antigen) = eukaryotic β-clamp = processivity factor for DNA Pol δ/ε PCNA encircles DNA and prevents polymerase dissociation → enables processive synthesis of thousands of nucleotides without falling off. Used as a histological marker of proliferating cells.
🟡 Cockayne Syndrome = TC-NER deficiency → premature ageing WITHOUT increased cancer risk Because GG-NER is intact in CS (only TC-NER is defective), the overall mutagenic burden is not dramatically increased, so cancer rates are not elevated. Contrast with XP (both NER pathways affected) → extreme cancer risk.
🟡 Frameshift mutations from indels (not multiples of 3) completely alter the downstream reading frame A 1 or 2 nucleotide insertion or deletion throws off the entire codon reading frame downstream → scrambled amino acid sequence → usually premature stop → nonfunctional protein. The most dramatic consequence of small indels.
🟡 The 4 kHz notch in audiometry corresponds to NIHL; the equivalent concept in genetics is mutational signatures — UV = C→T at TT sites, tobacco = G→T transversions Mutational signatures allow forensic identification of the carcinogen responsible for a tumour’s mutations — increasingly used in clinical oncology for treatment selection.
🟡 Telomerase is active in: germ cells, stem cells, cancer cells — suppressed in most somatic cells This explains why somatic cells age (progressive telomere shortening) while cancer cells do not. Telomerase reactivation is a hallmark of cancer. Anti-telomerase strategies are an active therapeutic research area.
🧠 Mnemonics & Memory Tricks
“PATEL” — DNA Replication Enzymes in Order P = Primase — synthesises RNA primer A = Anti-helicase / SSB proteins (Single Strand Binding proteins) — stabilise strands T = Topoisomerase — relaxes supercoiling E = Elongation by DNA polymerase (Pol III in prokaryotes; Pol δ/ε in eukaryotes) L = Ligase — seals the nicks
💡 Pro tip: This gives the key enzymes of replication in logical order. When any question asks “what enzyme does X in replication?” — run through PATEL and match.
“BNM-HR” — DNA Repair Pathways (in order of lesion size) B = BER (Base Excision Repair) — small, non-distorting lesions (uracil, 8-oxoG, AP sites) N = NER (Nucleotide Excision Repair) — large, helix-distorting lesions (thymine dimers, bulky adducts) M = MMR (Mismatch Repair) — replication mismatches and microsatellite errors H = HR (Homologous Recombination) — DSBs in S/G2 phase; error-free R = NHEJ (Rapid, error-prone) — DSBs in any phase; error-prone
💡 Pro tip: Repair pathways match the lesion type. Know BER → XP, Cockayne, Lynch, Fanconi, and AT are the 5 major DNA repair diseases. Each maps to one pathway.
“FAT LUNG” — Diseases of DNA Repair Deficiency F = Fanconi Anaemia (HR — interstrand crosslink repair) A = Ataxia-Telangiectasia (ATM kinase — DSB signalling) T = Trichothiodystrophy (NER — XPD/XPB) L = Lynch Syndrome (MMR — MLH1, MSH2, MSH6, PMS2) U = mucosa? — (MUTYH — BER) N = Nijmegen Breakage Syndrome (NBS1 — HR/DSB signalling) G = Global repair — XP (XPA-XPG, NER)
💡 Pro tip: Each disease has a signature that connects to its repair pathway. Know the clinical triad + the pathway + the gene.
“STOP TRAFFIC” — Features of p53 (The Guardian of the Genome) S = Stabilised by phosphorylation (from ATM/ATR kinases) → prevents MDM2 binding/ubiquitination T = Transcription factor → activates target genes O = Oxidative stress and oncogene activation → p53 activated P = p21 (CDK inhibitor) → cell cycle arrest (G1) T = TP53 mutated in >50% of all cancers R = RIBO (ribosomal stress) also activates p53 A = Apoptosis genes (BAX, PUMA, NOXA) induced F = Faulty DNA → p53 triggers arrest or apoptosis F = FAX Li-Fraumeni (Familial Li-Fraumeni): TP53 germline mutations I = Inhibited by MDM2 (E3 ubiquitin ligase → degrades p53) C = Cancer treatment target (MDM2 inhibitors, PRIMA-1 to restore mutant p53 function)
⚠️ Common Mistakes Students Make on This Topic
❌ Mistake: “DNA polymerase can start a new chain de novo — it just needs template DNA”
✅ Reality: DNA polymerase CANNOT initiate synthesis de novo. It can ONLY extend an existing 3′-OH group. This is why primase (RNA polymerase) is needed to synthesise a short RNA primer to provide the 3′-OH for DNA pol to extend. This is a fundamental property of all known DNA polymerases. 📝 How this gets tested: “Why is an RNA primer needed in DNA replication?” — The answer is specifically because DNA polymerase cannot initiate synthesis; it requires a free 3′-OH to add nucleotides to.
❌ Mistake: “Xeroderma Pigmentosum and Cockayne Syndrome are both characterised by increased skin cancer risk”
✅ Reality: XP has massively elevated skin cancer risk (1000× normal) because BOTH GG-NER (global genome) and TC-NER (transcription-coupled) are defective. Cockayne Syndrome has defective TC-NER ONLY; GG-NER remains functional → the global genome is still scanned and repaired → cancer rates are NOT significantly elevated. CS patients have premature ageing and neurological disease but not skin cancer predisposition. 📝 How this gets tested: “Which NER disease is associated with premature ageing but NOT with increased cancer?” — Cockayne Syndrome. “Which NER disease has 1000× increased skin cancer risk?” — Xeroderma Pigmentosum.
❌ Mistake: “Homologous recombination and NHEJ both repair DSBs with equal fidelity”
✅ Reality: HR is error-FREE (uses the sister chromatid as template to restore the exact sequence). NHEJ is error-PRONE (simply joins the broken ends, often with loss of 1–10 nucleotides → small deletions or insertions at the junction). HR can only operate in S/G2 (when sister chromatid is available). NHEJ operates throughout the cell cycle (G1, S, G2). 📝 How this gets tested: “A cell in G1 phase sustains a double-strand break — which repair pathway is primarily used?” = NHEJ. “Which repair pathway is error-free for DSBs?” = Homologous Recombination.
❌ Mistake: “A missense mutation always causes disease”
✅ Reality: A missense mutation changes one amino acid — the effect depends entirely on WHICH amino acid is changed and WHERE in the protein it is. A missense mutation at a critical active site will be catastrophic; one at a surface-exposed loop far from function may have no effect. Sickle cell (Glu→Val at position 6) is disease-causing; haemoglobin Korle Bu (Asp→Asn at position 73) has no clinical significance. Conservative substitutions (similar amino acids) are generally less harmful than non-conservative ones. 📝 How this gets tested: “A patient has a missense mutation — will this cause disease?” — Not necessarily; the answer is “it depends on the location and the amino acids involved.” This principle is critical for understanding variant of uncertain significance (VUS) classification in clinical genetics.
❌ Mistake: “Lynch syndrome is caused by the APC gene” ✅ Reality: Lynch syndrome is caused by MMR gene mutations (MLH1, MSH2, MSH6, PMS2). APC mutations cause Familial Adenomatous Polyposis (FAP) — a completely different colorectal cancer syndrome with hundreds/thousands of polyps. The two are easily confused because both are hereditary CRC syndromes, but their molecular mechanisms, clinical features, and clinical management are entirely different. Lynch syndrome has relatively few colorectal polyps but high cancer risk; FAP has hundreds/thousands of polyps. 📝 How this gets tested: “Which gene is most commonly mutated in Lynch syndrome?” = MLH1. “Which syndrome is caused by APC mutations?” = FAP. These are two of the most commonly tested genetics facts in oncology.
📝 5 Practice MCQs — Test Yourself Now
Q1: During DNA replication, one strand is synthesised continuously while the other is synthesised in short fragments. What is the enzymatic activity responsible for removing the RNA primers from the fragments on the lagging strand in prokaryotes?
- A. DNA Polymerase III 5’→3′ exonuclease
- B. DNA Polymerase I 5’→3′ exonuclease
- C. Primase 3’→5′ exonuclease
- D. DNA Ligase hydrolase activity
✅ Answer: B. DNA Polymerase I 5’→3′ exonuclease
Why correct: In prokaryotes, after DNA Pol III has synthesised each Okazaki fragment and extended to the 5′ end of the preceding RNA primer, DNA Polymerase I takes over. DNA Pol I has a unique 5’→3′ exonuclease activity (also called nick translation activity) that simultaneously removes the RNA primer in the 5’→3′ direction while its polymerase activity fills in the resulting gap with DNA. This makes DNA Pol I uniquely suited for this task — it both degrades (removes the primer) and synthesises (fills the gap) in one coordinated process. Finally, DNA Ligase seals the remaining nick.
Why A is wrong: DNA Pol III has 3’→5′ exonuclease activity (proofreading), not 5’→3′ exonuclease. Its 5’→3′ exonuclease activity is absent or negligible. It is the main replicative polymerase, not involved in primer removal. Why C is wrong: Primase SYNTHESISES RNA primers — it does not remove them. Primase has no exonuclease activity. Why D is wrong: DNA Ligase seals phosphodiester bond nicks. It has no exonuclease activity. Its role is the FINAL step — after Pol I has removed the primer and filled the gap.
Exam tip: In eukaryotes, primer removal is performed by the combined action of RNase H and FEN1 (Flap Endonuclease 1) — not by a DNA polymerase. The eukaryotic/prokaryotic distinction in primer removal mechanism is tested.
Q2: A 6-year-old child presents with severe sunburns after minimal UV exposure. Over the following years, she develops multiple basal cell carcinomas and squamous cell carcinomas on sun-exposed areas. Skin biopsy shows pyrimidine dimers that are not being repaired. Which protein is most likely deficient, and what repair pathway is impaired?
- A. MSH2; Mismatch Repair (MMR)
- B. XPC; Global Genome Nucleotide Excision Repair (GG-NER)
- C. CSB (ERCC6); Transcription-Coupled NER (TC-NER)
- D. PARP1; Base Excision Repair (BER)
✅ Answer: B. XPC; Global Genome Nucleotide Excision Repair (GG-NER)
Why correct: This is xeroderma pigmentosum (XP). The hallmarks: childhood onset photosensitivity, severe sunburn reactions, multiple skin malignancies at a young age, and persistent unrepaired pyrimidine dimers (thymine dimers). Pyrimidine dimers are helix-distorting lesions repaired by NER. XPC is the damage-recognition protein for Global Genome NER — it recognises the helix distortion caused by thymine dimers and initiates the repair cascade. Mutations in any of the XP genes (XPA–XPG) cause XP. XPC mutations are one of the most common forms. The 1000× elevated skin cancer risk results from accumulating unrepaired UV-induced mutations in tumour suppressor genes and proto-oncogenes.
Why A is wrong: MSH2 is an MMR protein — it recognises replication mismatches, not UV-induced pyrimidine dimers. MMR deficiency causes Lynch syndrome (colorectal and endometrial cancers), not childhood skin cancer from photosensitivity. Why C is wrong: CSB (ERCC6) deficiency causes Cockayne Syndrome — TC-NER is defective but GG-NER is intact. CS patients do NOT have elevated skin cancer rates (GG-NER repairs the genome as a whole). CS presents with premature ageing and neurological disease, not skin malignancies. Why D is wrong: PARP1 deficiency would impair BER — the pathway for small, non-helix-distorting lesions like uracil, 8-oxoguanine, and AP sites. Thymine dimers are large helix-distorting lesions repaired by NER, not BER.
Exam tip: XP = NER + skin cancers + photosensitvity. Cockayne Syndrome = TC-NER only + premature ageing + NO skin cancers. The distinction between these two is a classic exam discriminator.
Q3: A 45-year-old man presents with colorectal cancer in the right colon. Tumour immunohistochemistry shows absent staining for MLH1 and PMS2 proteins. PCR microsatellite analysis shows instability at 4 of 5 tested loci (MSI-High). His father and sister also had colorectal cancer before age 50. What is the most likely diagnosis, and what is the direct molecular mechanism linking the MMR defect to cancer development?
- A. Familial Adenomatous Polyposis (FAP); APC inactivation → constitutive Wnt signalling
- B. Lynch Syndrome; loss of MMR → microsatellite instability → frameshift mutations in tumour suppressor genes (e.g., TGFBR2, BAX, MLH3)
- C. MUTYH-Associated Polyposis; failure to correct 8-oxoguanine → G:C → T:A transversions in APC
- D. Serrated polyposis; BRAF V600E → MLH1 promoter methylation → sporadic MSI
✅ Answer: B. Lynch Syndrome; loss of MMR → microsatellite instability → frameshift mutations in tumour suppressor genes
Why correct: This is Lynch Syndrome (Hereditary Non-Polyposis CRC). The key features: MSI-High tumour, absent MLH1 and PMS2 on IHC (PMS2 is the obligate heterodimer partner of MLH1 and is lost when MLH1 is absent), right-sided CRC, family history meeting Amsterdam criteria (2 first-degree relatives with Lynch-associated cancers, one before 50, across 2 generations). The molecular mechanism: MMR loss → replication errors in repetitive microsatellite sequences accumulate → frameshift mutations in coding microsatellites of tumour suppressor genes (TGFBR2 is most commonly affected — frameshift in its A8 repeat causes loss of TGF-β growth inhibition in colorectal cancer; BAX frameshifts → anti-apoptotic; MLH3, MSH3 also affected) → clonal expansion → cancer. MSI-H Lynch syndrome tumours respond to pembrolizumab (anti-PD1).
Why A is wrong: FAP is caused by germline APC mutations with hundreds/thousands of adenomatous polyps; the tumour would have APC loss, not MMR deficiency, and IHC would show retained MMR protein expression. FAP is not MSI-H. Why C is wrong: MUTYH-associated polyposis is autosomal recessive; it causes multiple adenomas (not as many as FAP) and is associated with G:C → T:A transversions (from failure to correct 8-oxoG lesions), not MSI. IHC would show retained MMR proteins. Why D is wrong: Sporadic MSI-H CRC from MLH1 promoter hypermethylation is the most common mechanism of MSI-H CRC overall — but it is NOT hereditary. Sporadic MLH1 hypermethylation is typically associated with BRAF V600E mutation, older women, right-sided cancer — but there would be NO family history consistent with Lynch syndrome. The family history here (father and sister with CRC <50) is the key indicator of hereditary rather than sporadic disease.
Exam tip: Lynch syndrome work-up: CRC → tumour testing (MSI-PCR + IHC) → if dMMR/MSI-H → germline MMR testing. MSI-H tumours → pembrolizumab (FDA tumour-agnostic approval). The MLH1/PMS2 vs MSH2/MSH6 loss pattern on IHC indicates which gene is primarily affected.
Q4: A patient with advanced ovarian cancer and a known germline BRCA2 mutation has progressed on platinum-based chemotherapy. She is started on olaparib. The oncologist explains that olaparib exploits “synthetic lethality.” Which explanation best describes this mechanism?
- A. Olaparib inhibits DNA Pol α, blocking primer synthesis — in BRCA2-deficient cells, no alternative primer synthesis exists
- B. BRCA2-deficient cells cannot perform homologous recombination (HR). Olaparib inhibits PARP, preventing repair of single-strand breaks. Stalled replication forks collapse to double-strand breaks that BRCA2-deficient cells cannot repair by HR → cell death selectively in tumour cells
- C. Olaparib directly alkylates BRCA2-mutant DNA with greater efficiency than normal DNA, causing tumour-selective toxicity
- D. Olaparib activates ATM kinase only in BRCA2-deficient cells, triggering p53-dependent apoptosis selectively in tumour cells
✅ Answer: B. BRCA2-deficient cells cannot perform homologous recombination (HR). Olaparib inhibits PARP, preventing SSB repair → replication fork collapse → DSBs → cell death selectively in tumour cells
Why correct: This is the synthetic lethality principle. Normal cells can survive either BRCA2 loss OR PARP inhibition individually (because alternative repair pathways compensate). However, when BOTH are lost simultaneously — BRCA2 (HR) deficiency + PARP inhibition (SSB repair) — the combination is lethal to cells:
- PARP detects and repairs single-strand breaks via BER
- When PARP is inhibited, SSBs persist
- Replication forks encounter SSBs → fork collapse → DSBs
- Normal cells: HR (using BRCA2) repairs the DSBs → survival
- BRCA2-deficient tumour cells: Cannot perform HR → DSBs accumulate → cell death This creates tumour-selective killing — somatic cells heterozygous for BRCA2 (one functional copy) can still perform HR; only the tumour cells with biallelic BRCA2 loss are selectively killed.
Why A is wrong: Olaparib has no effect on DNA Pol α or primer synthesis. Its exclusive target is PARP1 and PARP2 (ADP-ribosylation of proteins at sites of SSBs). Why C is wrong: Olaparib does not alkylate DNA. It is not a DNA-damaging agent directly — it works by preventing repair of existing DNA damage. Why D is wrong: Olaparib does not directly activate ATM kinase. ATM is activated by the DSBs that result from collapsed replication forks — but this is a downstream consequence of PARP inhibition, not a direct target of olaparib.
Exam tip: PARP inhibitor synthetic lethality: BRCA1/2 deficiency (no HR) + PARP inhibition (no SSB repair) = synthetic lethality. Approved PARP inhibitors: olaparib, rucaparib, niraparib, talazoparib. BRCA1/2-deficient ovarian, breast, prostate, and pancreatic cancers are the primary indications.
Q5: During S phase, replication of a eukaryotic chromosome requires the coordinated action of multiple proteins at the replication fork. Which of the following correctly pairs a protein with its specific function?
- A. PCNA — unwinds the double helix ahead of the fork; MCM2-7 — provides processivity to DNA polymerase
- B. Primase — synthesises the RNA primer providing a 3′-OH for extension; FEN1 — removes RNA primer flaps during Okazaki fragment maturation
- C. DNA Pol γ — replicates nuclear chromosomal DNA; PCNA — repairs mitochondrial DNA
- D. Topoisomerase I — introduces negative supercoils to drive replication; RPA — removes RNA primers
✅ Answer: B. Primase — synthesises the RNA primer; FEN1 — removes RNA primer flaps
Why correct: This correctly pairs the protein with its actual function. Primase is part of the primosome complex (with DNA Pol α in eukaryotes) and synthesises short RNA primers (8–10 nt) on both the leading and lagging strand template to provide the essential 3′-OH group that DNA polymerase can then extend. FEN1 (Flap Endonuclease 1) is the eukaryotic enzyme responsible for removing the displaced RNA primer flap that forms when DNA Pol δ extends a new Okazaki fragment and encounters and displaces the 5′ end of the preceding primer → creating a 5′ flap → FEN1 cleaves the flap → gap filled by Pol δ → ligated by Ligase I. Both functions are correctly assigned.
Why A is wrong: PCNA does NOT unwind the double helix. The MCM2-7 complex is the replicative helicase (unwinds DNA). PCNA is the sliding clamp (β-clamp equivalent in eukaryotes) that provides processivity to DNA Pol δ/ε. The proteins are swapped in this option. Why C is wrong: DNA Pol γ replicates MITOCHONDRIAL DNA (not nuclear). Nuclear chromosomal DNA is replicated by DNA Pol δ (lagging) and Pol ε (leading). PCNA is involved in nuclear DNA replication, not mitochondrial — mitochondrial replication uses its own processivity factors. Why D is wrong: Topoisomerase I RELAXES supercoils — it relieves positive supercoiling ahead of the fork by cutting ONE strand, allowing rotation, then resealing. It does NOT introduce negative supercoils (that is DNA gyrase/Topo II in prokaryotes). RPA is a single-strand binding protein — it does NOT remove RNA primers; it protects ssDNA and helps load repair/replication proteins.
Exam tip: Know the specific functions of all major replication proteins: helicase (MCM2-7) = unwinds; SSB/RPA = stabilises ssDNA; Topoisomerase = relieves supercoiling; Primase = RNA primer; PCNA = processivity clamp; Pol δ/ε = synthesis; FEN1 = flap removal; Ligase I = nicks sealed. Any exam question on replication will pair protein with function — know each one.
📚 References
📖 Harper’s Illustrated Biochemistry — Rodwell et al. | Chapter 35: DNA Organisation, Replication & Repair; Chapter 36: RNA Synthesis, Processing & Modification
📖 Lippincott’s Illustrated Reviews: Biochemistry — Ferrier | Chapter 29: DNA Structure and Replication; Chapter 30: DNA Repair and Mutation
📖 Lehninger Principles of Biochemistry — Nelson & Cox | Chapter 24: Genes and Chromosomes; Chapter 25: DNA Metabolism
📖 Stryer’s Biochemistry — Berg, Tymoczko & Stryer | Chapter 28: DNA Replication, Repair and Recombination
📖 Robbins Basic Pathology — Kumar et al. | Chapter 7: Genetic and Pediatric Diseases (Cancer genetics)
📖 Alberts et al. — Molecular Biology of the Cell | Chapter 5: DNA, Chromosomes and Genomes; Chapter 6: DNA Replication
🚀 Keep Practising — You Are Not Done Yet
DNA questions in NEET PG and USMLE span from pure biochemistry (replication enzymes, repair pathways) through clinical genetics (Lynch syndrome, XP, Fanconi anaemia, BRCA1/2) to pharmacology (fluoroquinolones, cisplatin, PARP inhibitors). It is one of the few topics where a single conceptual framework connects all these seemingly different areas.
The students who score highest on DNA questions are those who can link mechanism to disease to drug. Why does XP cause skin cancer? → NER deficiency → unrepaired thymine dimers → mutations. Why do PARP inhibitors work in BRCA-mutant cancers? → Synthetic lethality between HR and SSB repair. Why does ciprofloxacin kill bacteria? → DNA gyrase inhibition → DSBs → bactericidal.
medicalmcq.in has free Biochemistry and Molecular Biology MCQs covering DNA replication, repair pathways, mutations, and pharmacology — all in clinical-scenario format with mechanistic explanations.