What You Will Learn in This Article
- Trace the complete biosynthesis of cortisol from cholesterol through every enzymatic step
- Name every enzyme in the steroidogenesis pathway and identify which ones are deficient in congenital adrenal hyperplasia (CAH)
- Explain the HPA axis — how CRH, ACTH, and cortisol are regulated and what feedback loops control them
- List all the major physiological actions of cortisol across metabolism, immunity, cardiovascular, bone, and CNS
- Describe how cortisol is transported, metabolised, and excreted
- Connect cortisol excess (Cushing syndrome) and deficiency (Addison disease, CAH) to their clinical presentations and investigations
- Explain the pharmacology of synthetic glucocorticoids — relative potencies, side effects, and clinical uses
📖 Introduction: Why This Topic Matters in Exams
A 34-year-old woman presents with weight gain concentrated around her trunk and face, purple striae on her abdomen, thin fragile skin, and new-onset hypertension. Her random cortisol is elevated, and her cortisol does not suppress with low-dose dexamethasone. She is found to have a pituitary adenoma secreting ACTH. This is Cushing disease — the most common cause of endogenous Cushing syndrome — and every feature of her presentation is a direct consequence of chronic cortisol excess. Understanding cortisol is not abstract endocrinology; it is the molecular explanation for some of the most recognisable syndromes in clinical medicine.
Cortisol appears in virtually every major entrance exam, and it does so across disciplines. In biochemistry, the steroidogenesis pathway and enzyme deficiencies are direct recall targets. In physiology, the HPA axis regulation, diurnal variation, and stress response are core. In endocrinology, Cushing syndrome, Addison disease, and congenital adrenal hyperplasia (CAH) are among the highest-yield clinical conditions. In pharmacology, the synthetic glucocorticoids — prednisolone, dexamethasone, hydrocortisone — are tested for their relative potencies, durations, and side effects.
This article covers the full scope systematically: biosynthesis from cholesterol to cortisol, HPA axis regulation, all physiological actions, transport and metabolism, clinical disorders, and pharmacology. By the end, you will be able to handle any exam question on cortisol — biochemical, physiological, or clinical.
🔬 Section 1 — Cholesterol to Cortisol: The Steroidogenesis Pathway
Overview: The Steroid Hormone Family
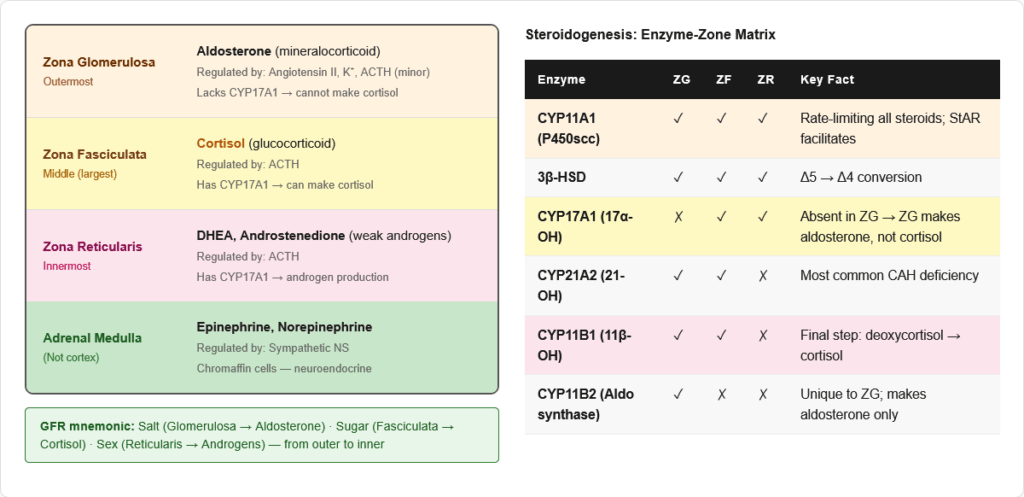
All steroid hormones are derived from cholesterol (C27). The adrenal cortex is divided into three zones, each producing different steroids:
| Zone | Hormone Produced | Mnemonic |
|---|---|---|
| Zona Glomerulosa (outer) | Aldosterone (mineralocorticoid) | “GFR” — Glomerulosa/Fasciculata/Reticularis |
| Zona Fasciculata (middle) | Cortisol (glucocorticoid) | |
| Zona Reticularis (inner) | DHEA, androstenedione (androgens) |
Memory trick: “GFR from outside in — Salt (Glomerulosa), Sugar (Fasciculata), Sex (Reticularis)”
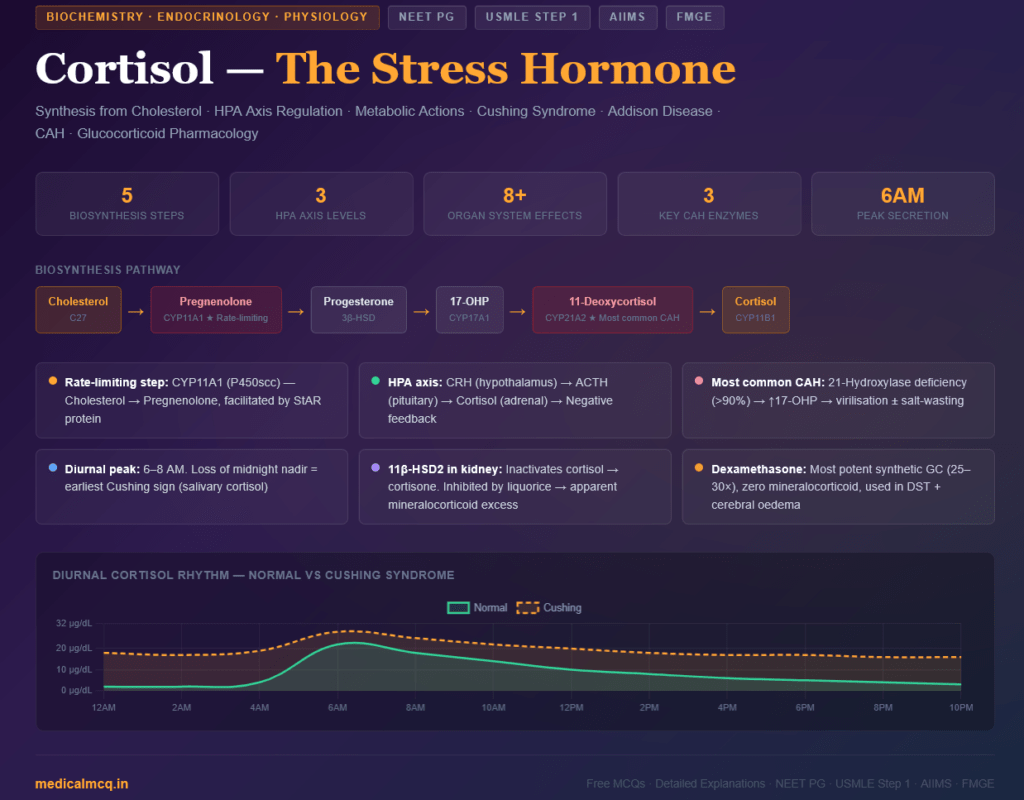
The Enzymatic Steps: Cholesterol → Cortisol
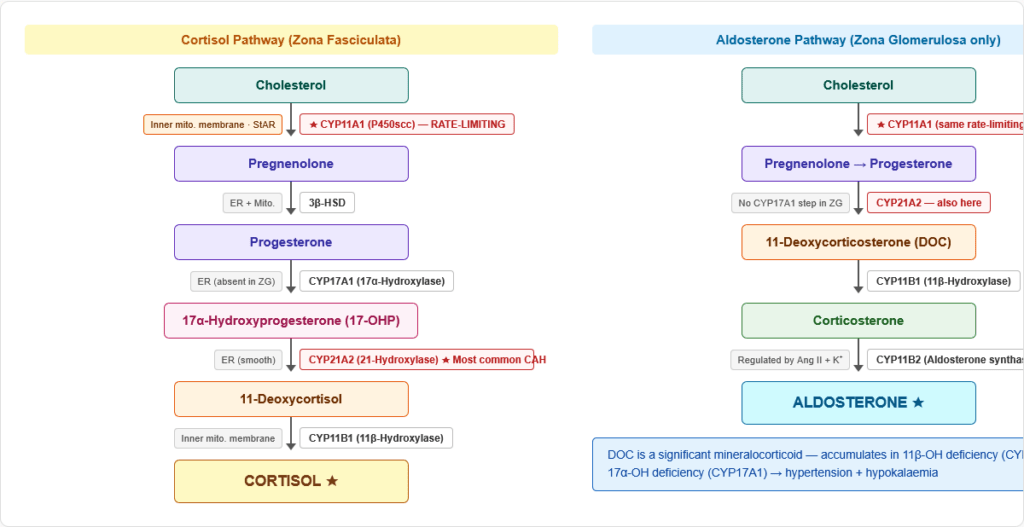
Step 1 — Cholesterol → Pregnenolone
- Enzyme: Cholesterol side-chain cleavage enzyme (CYP11A1 / P450scc)
- Location: Inner mitochondrial membrane
- Cofactors: NADPH, O₂
- Rate-limiting step of ALL steroid hormone synthesis
- Cholesterol transport into mitochondria is facilitated by StAR (Steroidogenic Acute Regulatory protein) — the acute regulatory protein that allows rapid increases in steroid output in response to ACTH
- ACTH stimulates StAR expression and CYP11A1 activity — this is how ACTH acutely increases cortisol production
Step 2 — Pregnenolone → Progesterone
- Enzyme: 3β-Hydroxysteroid dehydrogenase (3β-HSD) — type 2 in adrenal
- Location: ER and mitochondria
- Converts Δ5 steroids (double bond at C5) to Δ4 steroids (double bond at C4)
- Deficiency: 3β-HSD deficiency → impairs cortisol, aldosterone, AND androgen synthesis
Step 3 — Progesterone → 17α-Hydroxyprogesterone (17-OHP)
- Enzyme: 17α-Hydroxylase (CYP17A1)
- Location: ER (smooth)
- This step is absent in Zona Glomerulosa (which lacks CYP17A1) → explains why ZG makes aldosterone but not cortisol
- Most important step for CAH diagnosis: CYP17A1 deficiency → cannot make cortisol or sex steroids but CAN make aldosterone → hypertension + hypogonadism
Step 4 — 17-OHP → 11-Deoxycortisol
- Enzyme: 21-Hydroxylase (CYP21A2)
- Location: ER
- Most commonly deficient enzyme in CAH (>90% of cases)
- 21-Hydroxylase deficiency → 17-OHP accumulates → shunted to androgen pathway → virilisation
- Also affects aldosterone synthesis (Progesterone → 11-deoxycorticosterone step) → salt wasting
Step 5 — 11-Deoxycortisol → Cortisol
- Enzyme: 11β-Hydroxylase (CYP11B1)
- Location: Mitochondria (inner membrane)
- Second most common cause of CAH (~5% of cases)
- 11β-Hydroxylase deficiency → 11-deoxycortisol and 11-deoxycorticosterone accumulate → DOC (a mineralocorticoid) causes hypertension + hypogonadism
The Parallel Pathway: Aldosterone Synthesis
After Progesterone → 11-deoxycorticosterone (by CYP21A2):
- 11-Deoxycorticosterone → Corticosterone (by CYP11B1)
- Corticosterone → Aldosterone (by CYP11B2 / Aldosterone synthase) — in Zona Glomerulosa only
- CYP11B2 is the enzyme unique to aldosterone synthesis (absent in ZF and ZR)
Summary: Key Enzymes and Their Zones
| Enzyme | Gene | Location in Cell | Zone | Deficiency Consequence |
|---|---|---|---|---|
| P450scc | CYP11A1 | Inner mito. membrane | All zones | No steroid hormones (lethal) |
| 3β-HSD | HSD3B2 | ER + mito. | All zones | ↓ all steroids + mild virilisation |
| 17α-Hydroxylase | CYP17A1 | ER | ZF + ZR (NOT ZG) | No cortisol/sex steroids; ↑ aldosterone → HTN + hypogonadism |
| 21-Hydroxylase | CYP21A2 | ER | ZF + ZG | Most common CAH; virilisation ± salt-wasting |
| 11β-Hydroxylase | CYP11B1 | Inner mito. membrane | ZF + ZG | 2nd most common CAH; HTN + virilisation |
| Aldosterone synthase | CYP11B2 | Inner mito. membrane | ZG only | Hypoaldosteronism (salt-wasting, no HTN) |
⚙️ Section 2 — The HPA Axis: Regulation of Cortisol Secretion
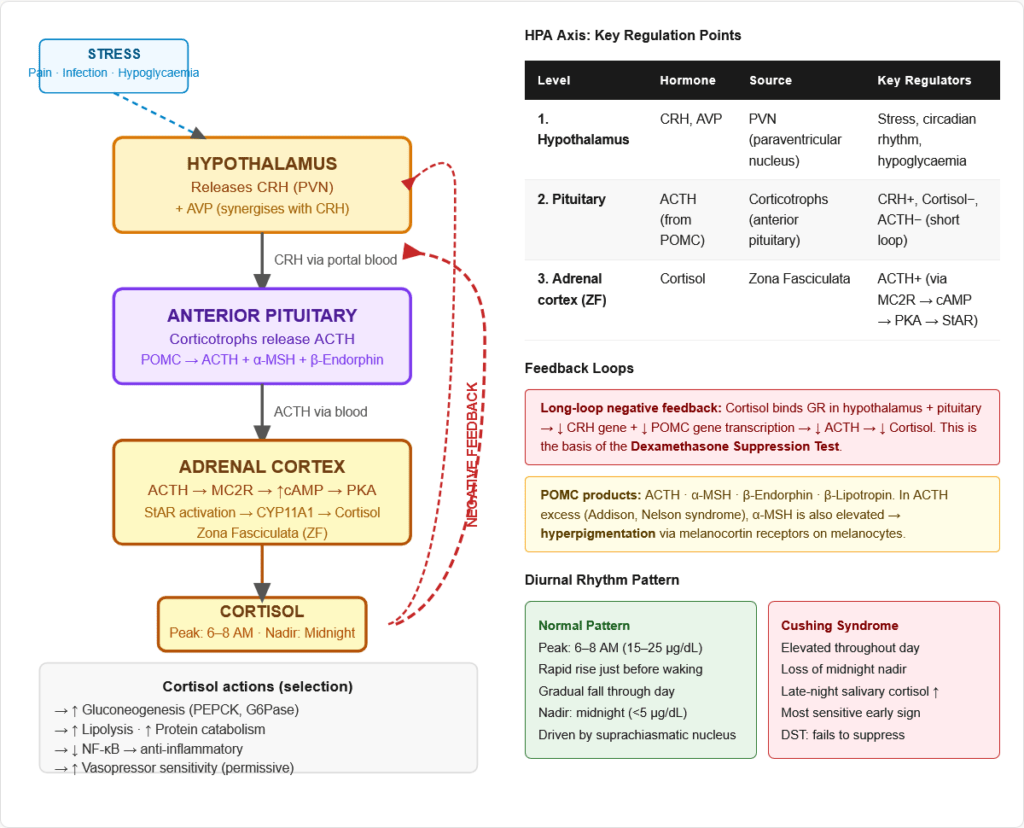
The Three-Level Hierarchy
Level 1: Hypothalamus
- Releases: Corticotropin-Releasing Hormone (CRH) from the paraventricular nucleus (PVN)
- CRH travels via the hypothalamo-pituitary portal system to the anterior pituitary
- Stimulated by: stress (physical, psychological, inflammatory), hypoglycaemia, exercise, sleep-wake cycle
- Also releases Arginine Vasopressin (AVP/ADH) which synergises with CRH to stimulate ACTH release
Level 2: Anterior Pituitary (Corticotrophs)
- CRH binds to corticotrophs → stimulates release of ACTH (Adrenocorticotropic hormone)
- ACTH is cleaved from a large precursor protein POMC (Pro-opiomelanocortin)
- POMC also gives rise to: β-Endorphin, α-MSH (melanocyte stimulating hormone), β-Lipotropin
- Why excess ACTH causes hyperpigmentation: ACTH and α-MSH share the same N-terminal sequence. In states of ACTH excess (Addison disease, Nelson syndrome), α-MSH is also elevated → stimulates melanocytes → hyperpigmentation of skin, buccal mucosa, scars
Level 3: Adrenal Cortex (Zona Fasciculata)
- ACTH binds to MC2R (melanocortin receptor 2) on adrenocortical cells → activates adenylyl cyclase → ↑ cAMP → activates PKA
- PKA actions: phosphorylates StAR (acute cortisol release), upregulates CYP11A1 and other steroidogenic enzymes (chronic), stimulates cholesterol ester hydrolase (releases free cholesterol)
Feedback Loops
Negative Feedback — Cortisol:
- Cortisol acts at BOTH hypothalamus (reduces CRH) AND anterior pituitary (reduces ACTH synthesis and release)
- Long-loop feedback: Cortisol binds glucocorticoid receptors (GR) in hypothalamus and pituitary → reduces CRH and POMC gene transcription
- This is the basis of the dexamethasone suppression test (DST): In normal people, exogenous dexamethasone (a potent GC that does not cross-react with cortisol assays) suppresses ACTH → cortisol falls. In Cushing syndrome, this suppression is blunted or absent.
Short-loop feedback:
- ACTH inhibits its own release from the pituitary (short loop)
Ultrashort-loop feedback:
- CRH inhibits its own release from the hypothalamus
Diurnal Rhythm (Circadian Variation)
Cortisol follows a strict circadian pattern driven by the suprachiasmatic nucleus:
- Peak: Early morning (6–8 AM) — highest cortisol, 15–25 µg/dL
- Nadir: Midnight — lowest cortisol, often <5 µg/dL
- The diurnal rhythm is lost in Cushing syndrome — midnight cortisol remains elevated
- Loss of diurnal rhythm is one of the earliest and most sensitive tests for Cushing syndrome (late-night salivary cortisol)
- Disrupted by: night-shift work, jet lag, depression, critical illness
Stress Response
Stress (physical injury, surgery, infection, haemorrhage, severe pain) causes immediate HPA activation:
- CRH surges → ACTH surges → cortisol can rise to 3–5× baseline within 15–30 minutes
- This “stress response” is essential for survival:
- Mobilises glucose (anti-insulin effects)
- Maintains vascular tone (permissive effect on catecholamines)
- Suppresses inflammation (prevents excessive immune damage)
- Redirects energy from growth/reproduction to immediate survival
🔬 Section 3 — Physiological Actions of Cortisol
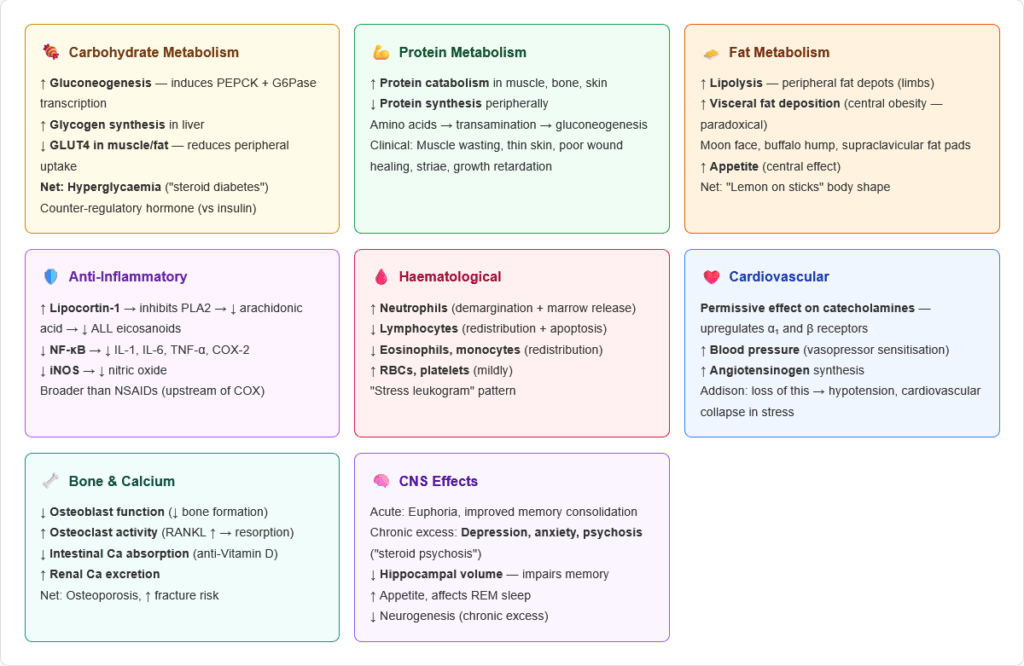
Cortisol has effects on virtually every organ system. Understanding WHY each effect occurs mechanistically is what separates exam-ready knowledge from rote memorisation.
Mechanism of Action — The Nuclear Receptor Pathway
Cortisol (being a lipophilic steroid) diffuses freely through cell membranes and binds to the glucocorticoid receptor (GR) in the cytoplasm. The GR is normally bound to HSP90 (heat shock protein 90) which keeps it inactive. When cortisol binds:
- Cortisol + GR → conformational change → HSP90 dissociates
- Cortisol-GR complex translocates to the nucleus
- Dimerises and binds to Glucocorticoid Response Elements (GREs) on DNA
- Recruits co-activators or co-repressors → alters gene transcription
- Net effect: induces or suppresses hundreds of genes
Transrepression: Cortisol-GR complex can also directly bind and inactivate transcription factors like NF-κB and AP-1 → suppresses inflammatory cytokine genes (IL-1, IL-6, TNF-α, COX-2). This is the basis of anti-inflammatory action.
1. Metabolic Effects
Carbohydrate metabolism:
- ↑ Gluconeogenesis (induces PEPCK, G6Pase transcription → increases hepatic glucose production)
- ↑ Glycogen synthesis in liver (paradoxically, while raising blood glucose overall)
- ↓ Peripheral glucose uptake (reduces GLUT4 translocation in muscle and fat)
- Net effect: Hyperglycaemia — “steroid diabetes”
- Cortisol is a counter-regulatory hormone alongside glucagon, epinephrine, and GH
Protein metabolism:
- ↑ Protein catabolism in muscle, skin, bone (provides amino acids for gluconeogenesis)
- ↓ Protein synthesis in peripheral tissues
- Amino acids released → transamination → gluconeogenic substrates
- Clinical consequences: Muscle wasting, thin skin, poor wound healing, growth retardation in children
Fat metabolism:
- Complex and paradoxical:
- ↑ Lipolysis in peripheral fat depots (limbs) → releases FFA for energy
- ↑ Fat deposition in central depots (abdomen, neck, face — “buffalo hump,” “moon face”) via enhanced adipogenesis in visceral fat under hyperinsulinaemia
- Net clinical picture: Central obesity with peripheral fat loss (“lemon on sticks” appearance)
- ↑ Appetite and food intake (central effect)
2. Anti-Inflammatory and Immunosuppressive Effects
These are the most important pharmacological effects of glucocorticoids:
Cellular effects:
- ↓ Neutrophil migration to sites of inflammation (reduces adhesion molecule expression)
- Causes neutrophilia in blood (demargination — releases neutrophils from vessel walls + bone marrow release)
- ↓ Lymphocytes, eosinophils, basophils, monocytes in blood (redistribution to lymphoid tissue + apoptosis)
- “Stress leukogram”: ↑ Neutrophils + ↓ Lymphocytes + ↓ Eosinophils
Molecular effects:
- ↓ NF-κB activity → ↓ IL-1, IL-2, IL-6, TNF-α, IFN-γ transcription
- ↓ COX-2 (cyclooxygenase-2) → ↓ prostaglandins, thromboxanes
- ↑ Lipocortin-1 (Annexin A1) → inhibits phospholipase A2 → ↓ arachidonic acid release → ↓ all eicosanoids
- ↓ iNOS (inducible nitric oxide synthase) → ↓ nitric oxide
- ↓ Histamine synthesis and release
Consequences of chronic GC excess:
- Increased susceptibility to infection (especially opportunistic — fungi, TB reactivation)
- Impaired wound healing
- Blunted fever response (cortisol suppresses IL-1 which drives fever)
3. Cardiovascular Effects
- Permissive effect on catecholamines: Cortisol upregulates α1-adrenergic receptors and β-adrenergic receptors on blood vessels and heart → enhances vasopressor response to norepinephrine and epinephrine
- ↑ Cardiac output and heart rate
- ↑ Blood pressure (multiple mechanisms: permissive catecholamine effect + mild mineralocorticoid effect at high concentrations binding MR + ↑ angiotensinogen synthesis)
- Addison disease: Loss of cortisol → loss of permissive catecholamine effect → hypotension, cardiovascular collapse in stress
4. Renal and Electrolyte Effects
- At physiological concentrations: Cortisol has weak mineralocorticoid activity (binds MR)
- Normally protected by 11β-HSD2 in the kidney, which converts cortisol to inactive cortisone (so cortisol cannot overwhelm MR in the kidney)
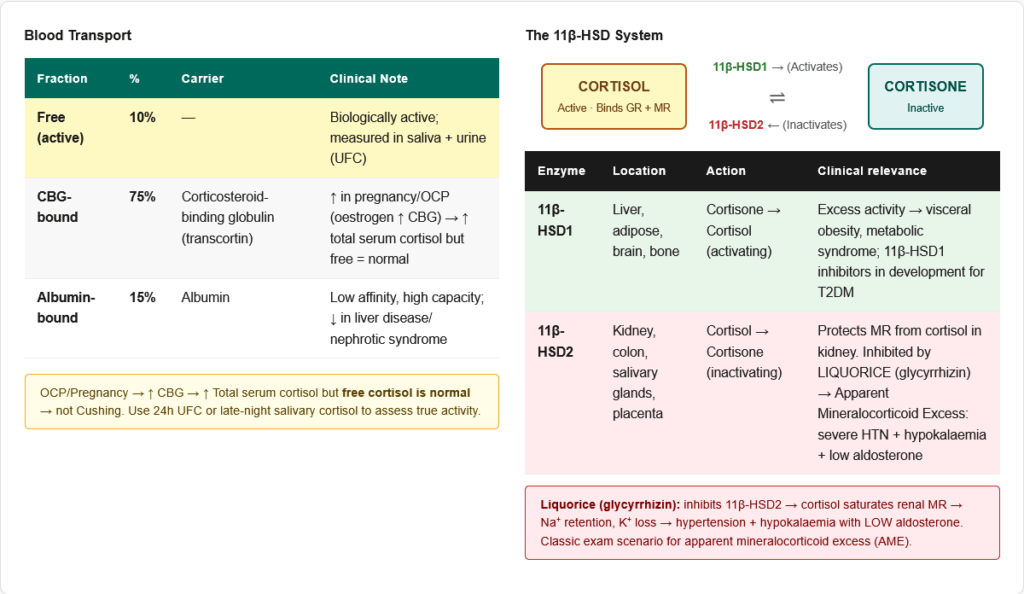
- At supraphysiological doses (Cushing syndrome): Overwhelms 11β-HSD2 → cortisol acts on MR → Na⁺ retention, K⁺ excretion → hypokalaemia, hypertension
- Apparent mineralocorticoid excess (AME): 11β-HSD2 deficiency → cortisol saturates renal MR → severe hypertension + hypokalaemia despite low/normal aldosterone. Caused by liquorice (glycyrrhizin inhibits 11β-HSD2).
5. Bone and Calcium Metabolism
Chronic excess cortisol is directly destructive to bone:
- ↓ Osteoblast function and number (reduces bone formation)
- ↑ Osteoclast activity (via RANKL upregulation → increased bone resorption)
- ↓ Intestinal calcium absorption (antagonises Vitamin D)
- ↑ Renal calcium excretion
- Net result: Osteoporosis — most common serious complication of chronic GC therapy
- ↑ PTH (secondary hyperparathyroidism from hypocalcaemia) → further bone resorption
- Avascular necrosis of femoral head is a particularly severe complication of high-dose GC therapy
6. Growth and Reproductive Effects
- Suppresses growth hormone (GH) axis: Reduces IGF-1 production → growth suppression in children
- Suppresses gonadotropins (LH, FSH): Reduces GnRH → hypogonadism, irregular menstruation, reduced fertility
- HPA-HPG cross-talk: Chronic stress/cortisol excess → reproductive suppression (physiologically appropriate — not a good time to reproduce during severe stress)
7. CNS Effects
- Mood effects: Acute cortisol → euphoria, increased energy. Chronic excess → depression, anxiety, psychosis (“steroid psychosis”)
- Cognitive effects: Moderate cortisol → improved memory consolidation. Excess → impairs hippocampal function, reduces neurogenesis, can cause memory deficits
- Appetite and sleep: Increases appetite, affects sleep architecture (reduces REM, can cause insomnia at high doses)
- Raises seizure threshold (short term) but can lower it chronically
8. Haematological Effects
| Cell Type | Effect of Cortisol | Mechanism |
|---|---|---|
| Neutrophils | ↑ in blood | Demargination + bone marrow release |
| Lymphocytes | ↓ in blood | Redistribution + apoptosis |
| Eosinophils | ↓ in blood | Redistribution to lymphoid tissue |
| Monocytes/Macrophages | ↓ in blood | Redistribution |
| Red blood cells | ↑ (mild) | ↑ Erythropoiesis via EPO |
| Platelets | ↑ | ↑ Thrombopoiesis |
🔬 Section 4 — Transport, Metabolism, and Excretion of Cortisol
Transport in Blood
- Cortisol-binding globulin (CBG / Transcortin): 75% of circulating cortisol — specifically binds cortisol with high affinity
- Albumin: 15% — low affinity, high capacity binding
- Free cortisol: ~10% — the biologically active fraction
- Total serum cortisol measures all forms; free cortisol (in urine or saliva) reflects biological activity
Clinical implications:
- Pregnancy / OCP use / hyperthyroidism: ↑ CBG synthesis (oestrogen stimulates liver CBG production) → ↑ total serum cortisol, but free cortisol is normal → no Cushing syndrome clinically
- Liver disease / nephrotic syndrome: ↓ CBG and albumin → ↓ total cortisol, but free cortisol may be normal
- Key distinction: Urinary free cortisol (UFC) and late-night salivary cortisol measure free cortisol → not affected by CBG changes
Metabolism and Inactivation
Cortisol is metabolised primarily in the liver:
Step 1: Cortisol ↔ Cortisone
- 11β-HSD1 (in liver, adipose, brain): Converts cortisone → cortisol (activating)
- 11β-HSD2 (in kidney, colon, salivary glands): Converts cortisol → cortisone (inactivating)
- This tissue-specific interconversion is critical for:
- Protecting kidney MR from cortisol (11β-HSD2)
- Providing active cortisol in adipose tissue (11β-HSD1 — relevant to metabolic syndrome)
Step 2: Reduction of the A-ring
- Cortisol → Dihydrocortisol → Tetrahydrocortisol (THF)
- Enzymes: 5β-reductase (predominantly) and 5α-reductase
Step 3: Conjugation
- Tetrahydrocortisol + UDP-glucuronic acid → Tetrahydrocortisol glucuronide
- Also sulphation (minor)
- Makes the steroid water-soluble for urinary excretion
Urinary Excretion
- 17-Hydroxycorticosteroids (17-OHCS): Measures THF, THF cortisone, and cortol — used in older cortisol assays
- Urinary Free Cortisol (UFC): Most sensitive test for cortisol excess; measures unbound cortisol filtered at glomerulus; 24-hour collection
- Urinary 17-Ketosteroids (17-KS): Measures adrenal androgens (DHEA, androstenedione) — NOT a good measure of cortisol itself (cortisol is a 21-carbon steroid, 17-KS are 19-carbon)
- Normal: 24-hour UFC = 20–90 µg/day; >300 µg/day strongly suggests Cushing syndrome
Half-life and Duration
- Plasma half-life of cortisol: ~60–90 minutes
- Biological effect duration: Several hours (due to nuclear receptor effects on gene transcription)
- Diurnal peak: 6–8 AM; nadir: midnight
🏥 Section 5 — Clinical Disorders of Cortisol
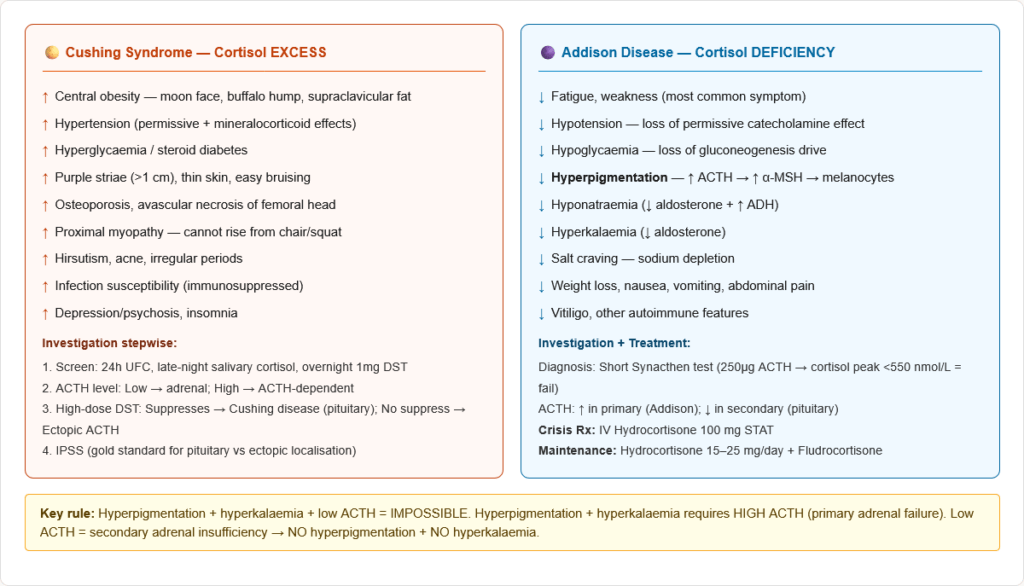
Cushing Syndrome — Cortisol Excess
Definition: Syndrome of chronic glucocorticoid excess from ANY cause.
Causes and Classification:
| Type | Cause | ACTH Level | Bilateral Adrenal? |
|---|---|---|---|
| Cushing Disease (most common endogenous) | Pituitary ACTH-secreting adenoma | High | Yes (bilateral hyperplasia) |
| Ectopic ACTH syndrome | ACTH from non-pituitary tumour (SCLC, carcinoid, MTC, phaeochromocytoma) | Very High | Yes (bilateral hyperplasia) |
| Adrenal adenoma | Autonomous cortisol secretion | Low (suppressed) | No (unilateral) |
| Adrenal carcinoma | Autonomous cortisol secretion | Low (suppressed) | No (unilateral) |
| Exogenous (most common overall) | Iatrogenic — oral/topical/inhaled/injected steroids | Low (suppressed) | Yes (bilateral atrophy) |
Clinical Features (Pathophysiology-linked):
| Feature | Mechanism |
|---|---|
| Central obesity (moon face, buffalo hump, supraclavicular fat) | ↑ Visceral adipogenesis, ↑ appetite |
| Muscle wasting (proximal myopathy) | ↑ Protein catabolism |
| Purple striae (>1 cm width) | Skin protein loss → thin dermis, ruptured dermal blood vessels |
| Easy bruising, thin skin | ↓ Collagen synthesis, capillary fragility |
| Hypertension | Permissive catecholamine effect + mild mineralocorticoid action |
| Hyperglycaemia / steroid diabetes | ↑ Gluconeogenesis, ↓ GLUT4 |
| Osteoporosis / fractures | ↓ Osteoblasts, ↑ osteoclasts, ↓ Ca absorption |
| Menstrual irregularity / hirsutism | ↑ Adrenal androgens + GnRH suppression |
| Hyperpigmentation | Only in ACTH-DEPENDENT causes (excess ACTH → α-MSH → melanocytes) |
| Immunosuppression | ↓ Lymphocytes, ↓ cytokines |
| Depression / psychosis | Hippocampal damage, disrupted neurotransmission |
| Hypokalaemia | Overwhelming MR at high cortisol (especially ectopic ACTH — extreme levels) |
Investigations for Cushing Syndrome:
Step 1 — Confirm cortisol excess (screening):
- 24-hour UFC (elevated)
- Late-night salivary cortisol (elevated — loss of nadir)
- Overnight 1 mg dexamethasone suppression test (cortisol fails to suppress to <50 nmol/L)
- Low-dose DST: 0.5 mg dexamethasone 6-hourly for 2 days → cortisol should suppress to <50 nmol/L in normals
Step 2 — Determine ACTH dependency:
- Plasma ACTH:
- ACTH undetectable/low (<10 pg/mL) → ACTH-independent (adrenal source)
- ACTH normal/elevated → ACTH-dependent (pituitary or ectopic)
Step 3 — If ACTH-dependent, localise:
- High-dose DST (2 mg dex 6-hourly for 2 days): Cortisol suppresses in Cushing disease (pituitary adenoma responds to high GC) but NOT in ectopic ACTH (autonomous secretion)
- Pituitary MRI
- Inferior Petrosal Sinus Sampling (IPSS): Gold standard for distinguishing pituitary from ectopic — CRH-stimulated ACTH gradient >2 (central:peripheral) = pituitary source
Addison Disease — Primary Adrenal Insufficiency
Causes:
- Autoimmune adrenalitis — most common in developed world (80%); associated with anti-21-hydroxylase antibodies, APS type 1 and 2
- Tuberculosis — most common worldwide; calcification on imaging
- Histoplasmosis, CMV, HIV
- Bilateral adrenal haemorrhage (Waterhouse-Friderichsen syndrome — meningococcal sepsis, DIC)
- Bilateral metastases (lung, breast, stomach, kidney cancers)
- Drugs: Ketoconazole (inhibits steroidogenesis), metyrapone, mitotane, rifampicin (accelerates GC metabolism)
Clinical Features:
- Fatigue, weakness (most common)
- Hyperpigmentation — pathognomonic of primary adrenal insufficiency with ACTH excess. Seen on sun-exposed areas, buccal mucosa, scars, pressure areas, genitalia, areolae
- Postural hypotension — loss of cortisol + aldosterone → Na⁺ loss, volume depletion, reduced vasopressor response
- Hyponatraemia — ADH release from volume depletion + aldosterone deficiency
- Hyperkalaemia — aldosterone deficiency
- Hypoglycaemia — loss of gluconeogenesis drive
- Weight loss, anorexia, nausea, vomiting, abdominal pain
- Salt craving (Na⁺ depletion)
- Vitiligo, other autoimmune features (if autoimmune cause)
Addisonian Crisis:
- Precipitated by: stress (infection, surgery, trauma) in a patient with adrenal insufficiency
- Features: Severe hypotension/shock, vomiting, abdominal pain, confusion, hypoglycaemia
- Treatment: IV hydrocortisone 100 mg bolus → 50–100 mg every 6–8 hours; IV 0.9% saline; glucose if hypoglycaemic
Investigations:
- Morning cortisol <100 nmol/L → likely deficiency; >550 nmol/L → excludes insufficiency
- Short Synacthen test: 250 µg ACTH IM/IV → cortisol measured at 30 and 60 minutes. Normal response: peak >550 nmol/L. A failure to respond confirms primary adrenal insufficiency.
- ACTH: Elevated in primary (Addison disease); low/undetectable in secondary (pituitary failure)
- Electrolytes: Hyponatraemia, hyperkalaemia (in primary)
- Anti-21-hydroxylase antibodies (autoimmune cause)
- Imaging: CT adrenals (enlargement/calcification/metastases)
Secondary vs Primary Adrenal Insufficiency:
| Feature | Primary (Addison) | Secondary (Pituitary/Hypothalamic) |
|---|---|---|
| ACTH | ↑ | ↓ |
| Hyperpigmentation | ✓ (↑ ACTH → ↑ α-MSH) | ✗ |
| Aldosterone | ↓ (primary—ZG also affected) | Normal (ACTH doesn’t regulate ZG) |
| Hyponatraemia | ✓ (aldosterone deficiency + ADH) | ✓ (SIADH-like, ADH ↑) |
| Hyperkalaemia | ✓ (aldosterone deficiency) | ✗ (aldosterone normal) |
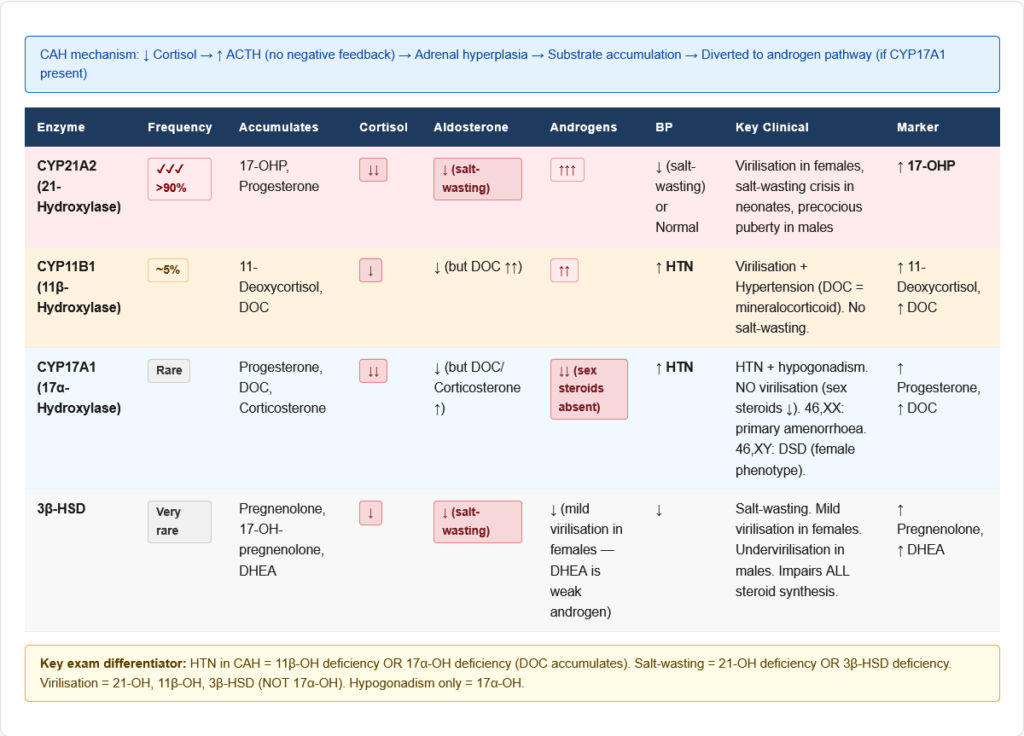
Congenital Adrenal Hyperplasia (CAH) — Enzyme Deficiencies
CAH is caused by deficiency of an enzyme in the cortisol synthesis pathway. Low cortisol → high ACTH (no negative feedback) → adrenal hyperplasia → excess intermediates diverted to androgen pathway.
21-Hydroxylase Deficiency (CYP21A2) — >90% of CAH cases
- Pathophysiology: 17-OHP accumulates → shunted to androgen synthesis → excess DHEA, androstenedione → testosterone
- Cortisol deficiency → high ACTH → adrenal hyperplasia
- Aldosterone synthesis also impaired (severe forms)
| Form | Severity | Features |
|---|---|---|
| Classic salt-wasting (most severe) | Complete enzyme loss | Virilisation + salt-wasting crisis (hyponatraemia, hyperkalaemia, hypotension) in neonate |
| Classic simple virilising | Partial enzyme function | Virilisation without salt-wasting |
| Non-classic (mild) | Mild enzyme deficiency | Premature puberty, hirsutism, irregular periods, mild virilisation — presents in childhood/adolescence |
- Diagnosis: Elevated 17-OHP (the accumulated substrate) — most specific marker; elevated androstenedione, testosterone
- Treatment: Hydrocortisone (replaces cortisol → suppresses ACTH → reduces androgen excess); Fludrocortisone (replaces aldosterone in salt-wasting form)
11β-Hydroxylase Deficiency (CYP11B1) — ~5% of CAH
- 11-Deoxycortisol and 11-deoxycorticosterone (DOC) accumulate
- DOC is a potent mineralocorticoid → hypertension + hypokalaemia
- Excess androgens → virilisation
- Key distinction from 21-OH deficiency: HTN (not salt-wasting) + virilisation
17α-Hydroxylase Deficiency (CYP17A1) — Rare
- Cannot make cortisol or sex steroids
- Excess progesterone → excess 11-deoxycorticosterone + corticosterone → hypertension + hypokalaemia
- Sex hormone deficiency: Female: primary amenorrhoea; Male: 46,XY DSD (male pseudohermaphroditism)
- No virilisation (because sex steroids are deficient, not in excess)
- HTN + hypogonadism is the key exam combination
🔬 Section 6 — Pharmacology: Synthetic Glucocorticoids
Relative Potencies
| Drug | Anti-inflammatory Potency | Mineralocorticoid Potency | Duration | Equivalent dose |
|---|---|---|---|---|
| Cortisol (hydrocortisone) | 1 (reference) | 1 | Short (8–12 h) | 20 mg |
| Prednisolone | 4 | 0.8 | Intermediate (12–36 h) | 5 mg |
| Methylprednisolone | 5 | 0.5 | Intermediate (12–36 h) | 4 mg |
| Triamcinolone | 5 | 0 | Intermediate | 4 mg |
| Dexamethasone | 25–30 | 0 (no mineralocorticoid) | Long (36–54 h) | 0.75 mg |
| Betamethasone | 25–30 | 0 | Long (36–54 h) | 0.6 mg |
| Fludrocortisone | 10 | 125 (high mineralocorticoid) | — | — |
| Beclometasone | Topical/inhaled | — | — | — |
Key facts for exams:
- Dexamethasone: Most potent anti-inflammatory synthetic GC, NO mineralocorticoid activity, longest duration, used in DST (does not interfere with cortisol assay), used for foetal lung maturation and cerebral oedema
- Fludrocortisone: Used specifically for mineralocorticoid replacement (Addison disease, salt-wasting CAH) — not for anti-inflammatory use
- Betamethasone/Dexamethasone: Given to mothers at 24–34 weeks gestation for foetal lung maturation (cross placenta well — not inactivated by placental 11β-HSD3; prednisolone is inactivated by placenta)
Clinical Uses of Glucocorticoids
Replacement therapy:
- Addison disease: Hydrocortisone 15–25 mg/day (morning-predominant dosing to mimic diurnal rhythm) + Fludrocortisone
- CAH: Hydrocortisone (suppresses ACTH excess) ± Fludrocortisone
Anti-inflammatory / Immunosuppressive:
- Rheumatoid arthritis, SLE, vasculitis, polymyalgia rheumatica, inflammatory bowel disease
- Asthma, COPD exacerbations, anaphylaxis
- Organ transplant rejection prevention
- Nephrotic syndrome (minimal change disease)
Other specific uses:
- Cerebral oedema (vasogenic — tumour, abscess): Dexamethasone
- Bacterial meningitis: Dexamethasone (reduces neurological sequelae by reducing inflammation)
- Foetal lung maturation: Betamethasone or dexamethasone IM to mother
- Spinal cord injury: Methylprednisolone (controversial)
- COVID-19: Dexamethasone (RECOVERY trial — reduced mortality in ventilated patients)
- Tumour lysis syndrome / hypercalcaemia of malignancy: Dexamethasone
- Immune thrombocytopaenia (ITP): Prednisolone
- Altitude sickness (cerebral/pulmonary oedema): Dexamethasone
Side Effects of Chronic Glucocorticoid Therapy
Metabolic:
- Hyperglycaemia / steroid-induced diabetes
- Hyperlipidaemia
- Weight gain, central obesity
Musculoskeletal:
- Osteoporosis (most common serious complication — bisphosphonates must be co-prescribed for long-term therapy)
- Proximal myopathy
- Avascular necrosis of femoral head (high-dose)
- Growth retardation in children
Gastrointestinal:
- Peptic ulceration (risk increased when combined with NSAIDs)
- Pancreatitis
Cardiovascular:
- Hypertension
- Fluid retention (sodium retention — especially older GCs with mineralocorticoid activity)
Immunological:
- Infections — bacterial, fungal, opportunistic (Pneumocystis jirovecii, aspergillosis)
- TB reactivation
- Impaired wound healing
Ophthalmological:
- Posterior subcapsular cataracts
- Glaucoma (raised IOP)
CNS:
- Steroid psychosis, depression, insomnia
- Behavioural changes in children
Endocrine:
- HPA axis suppression (if >3 weeks therapy → cannot stop abruptly — taper required)
- Cushing syndrome features
- Adrenal crisis if abruptly withdrawn
Skin:
- Atrophy, striae, easy bruising
- Acne
- Topical GC: skin atrophy, perioral dermatitis, steroid rosacea
🔄 Section 7 — Connections to Other Pathways and Systems
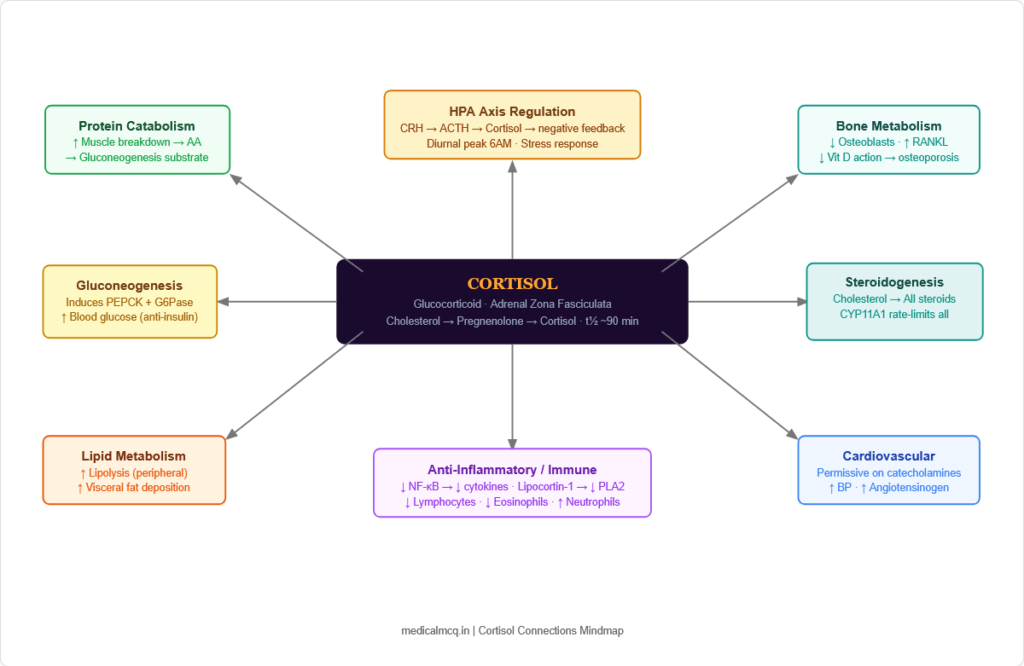
→ Cholesterol Metabolism — Cortisol synthesis begins with cholesterol. All steroid hormones (cortisol, aldosterone, testosterone, oestrogen, progesterone, vitamin D) share cholesterol as the common precursor. CYP11A1 (P450scc) — rate-limiting for ALL steroid synthesis — is the first committed step.
→ Gluconeogenesis — Cortisol directly induces PEPCK and G6Pase transcription → activates gluconeogenesis. This is why Cushing syndrome causes hyperglycaemia and why Addison disease causes hypoglycaemia. Cortisol also promotes protein catabolism → provides amino acid substrates for gluconeogenesis.
→ Lipid Metabolism — Cortisol activates hormone-sensitive lipase (HSL) in peripheral fat → lipolysis → free fatty acids for energy. It also promotes visceral adipogenesis under conditions of hyperinsulinaemia (paradoxical central fat deposition). Excess cortisol → hypertriglyceridaemia.
→ Amino Acid Metabolism — Cortisol is catabolic for muscle protein. Released amino acids (especially alanine) → liver → gluconeogenesis via the glucose-alanine cycle. This connects cortisol-driven proteolysis directly to hepatic glucose production.
→ Immune System / Eicosanoids — Cortisol induces lipocortin-1 (Annexin A1) → inhibits phospholipase A2 → blocks arachidonic acid release → reduces ALL eicosanoids (prostaglandins, leukotrienes, thromboxanes). This is the master upstream anti-inflammatory action that distinguishes GC from NSAIDs (which only inhibit COX).
→ HPA-HPG Cross-talk — Cortisol suppresses GnRH → LH/FSH → reduced gonadal steroid production. Chronic stress/Cushing syndrome → hypogonadism. Conversely, oestrogen (from OCP) ↑ CBG → ↑ total serum cortisol (not biologically active).
→ Bone Metabolism — Cortisol opposes vitamin D action on intestinal calcium absorption. Reduces osteocalcin (osteoblast marker). Increases RANKL → osteoclastogenesis. The combination makes GC-induced osteoporosis one of the most severe metabolic bone diseases.
🎯 High-Yield Exam Facts
🔴 Rate-limiting step of ALL steroid hormone synthesis: CYP11A1 (P450scc) — Cholesterol → Pregnenolone ACTH acutely stimulates this step via StAR protein. This is why ACTH is the master regulator of cortisol production.
🔴 Most common enzyme deficiency in CAH: 21-Hydroxylase (CYP21A2) — >90% of cases 17-OHP accumulates → excess androgens → virilisation ± salt-wasting. Diagnosis: Elevated 17-OHP.
🔴 Cortisol peak: 6–8 AM; Nadir: midnight. Loss of diurnal rhythm is the earliest sign of Cushing syndrome Late-night salivary cortisol is the most sensitive screening test for this reason.
🔴 ACTH excess → hyperpigmentation (through α-MSH from POMC). Seen in Addison disease and Nelson syndrome Hyperpigmentation is ONLY in ACTH-dependent states (primary adrenal insufficiency, ectopic ACTH) — NOT in adrenal adenoma or exogenous GC excess.
🔴 11β-HSD2 in kidney converts cortisol → cortisone, protecting MR from cortisol Inhibited by liquorice (glycyrrhizin) → apparent mineralocorticoid excess (AME) → severe hypertension + hypokalaemia. This is one of the most classically tested enzyme facts about cortisol metabolism.
🔴 Dexamethasone: most potent anti-inflammatory GC, ZERO mineralocorticoid activity, used in DST Used for: foetal lung maturation, cerebral oedema, bacterial meningitis, DST (does not interfere with cortisol assay), COVID-19.
🟠 11β-Hydroxylase deficiency (CYP11B1): virilisation + HYPERTENSION (DOC accumulates = mineralocorticoid) Contrast with 21-hydroxylase deficiency which causes salt-WASTING (no hypertension in classic forms). Both cause virilisation.
🟠 17α-Hydroxylase deficiency: no cortisol + no sex steroids + excess DOC → HTN + hypogonadism, NO virilisation The combination of hypertension + primary amenorrhoea (in 46,XX) or 46,XY DSD + no virilisation = 17α-hydroxylase deficiency.
🟠 Cortisol causes: ↑ neutrophils + ↓ lymphocytes + ↓ eosinophils = “stress leukogram” The classic WBC pattern of cortisol excess. Eosinopenia is one of the most specific haematological signs of acute cortisol excess.
🟠 GC-induced osteoporosis: first bisphosphonates must be co-prescribed for any patient on >3 months of systemic steroids Mechanism: ↓ osteoblasts, ↑ osteoclasts, ↓ intestinal Ca absorption, ↑ renal Ca loss, ↑ PTH (secondary hyperparathyroidism).
🟠 Addisonian crisis treatment: IV hydrocortisone 100 mg STAT — do not delay for investigations In suspected adrenal crisis: take blood for cortisol/ACTH, then give hydrocortisone immediately. Never delay treatment awaiting test results.
🟡 Pregnancy / OCP increases CBG → elevated TOTAL serum cortisol, but FREE cortisol is normal This is why pregnant women and OCP users appear to have high cortisol on standard tests — measure UFC or late-night salivary cortisol to assess true activity.
🟡 Betamethasone and dexamethasone cross the placenta (not inactivated by 11β-HSD3); prednisolone is inactivated This is why betamethasone (not prednisolone) is used for foetal lung maturation. Prednisolone is used when treating the mother (doesn’t reach foetus).
🟡 High-dose DST: Cushing disease suppresses (pituitary adenoma still has some GR); ectopic ACTH does NOT suppress This is the physiological basis of the high-dose DST used in Step 3 of Cushing syndrome workup.
🧠 Mnemonics & Memory Tricks
“GFR from outside in — Salt, Sugar, Sex” → Adrenal cortex zones and their products (outside → inside) G = Glomerulosa → Salt (Aldosterone — mineralocorticoid) F = Fasciculata → Sugar (Cortisol — glucocorticoid) R = Reticularis → Sex (Androgens — DHEA, androstenedione) 💡 Pro tip: Zone = Function = Hormone = “Salt-Sugar-Sex from outside in”
“17, 21, 11 — The Three CAH Enzymes to Know” → In order of importance/frequency in exams: 21-OH → Most common (>90%) → Virilisation ± Salt-wasting → Elevated 17-OHP 11β-OH → 2nd most common (~5%) → Virilisation + Hypertension (DOC excess) 17α-OH → Rare → Hypertension + Hypogonadism, NO virilisation Memory: “21 is most, 11 is second, 17 is third — but 17 is the one with no sex hormones” 💡 Pro tip: “Which CAH has hypertension?” — 11β and 17α (both have excess DOC/corticosterone) — NOT 21-hydroxylase deficiency.
“COLDER” — Features of Addison Disease C = Cortisol deficiency (hypoglycaemia, weakness) O = Other autoimmune (vitiligo, APS) L = Loss of aldosterone (hyponatraemia, hyperkalaemia, hypotension) D = Darkness (hyperpigmentation — elevated ACTH/α-MSH) E = Electrolytes abnormal (Na↓, K↑) R = Really needs hydrocortisone (replacement therapy) 💡 Pro tip: The key distinguishing feature of Addison disease vs secondary adrenal insufficiency is “DARK” (hyperpigmentation) + hyperkalaemia — both require ↑ ACTH which only occurs in primary disease.
“DEX is Best for Tests (and No Water Retention)” → Dexamethasone properties DEX = Dosen’t cause water retention (zero mineralocorticoid potency) is Best = highest anti-inflammatory potency (25–30× cortisol) for Tests = used for the Dexamethasone Suppression Test No Water = no mineral retention = used when you want pure GC effect without BP/fluid effects 💡 Pro tip: When a question asks “which GC is used for DST?” or “which GC has no mineralocorticoid effect?” — always dexamethasone.
⚠️ Common Mistakes Students Make on This Topic
❌ Mistake: “21-Hydroxylase deficiency always causes hypertension” ✅ Reality: 21-Hydroxylase deficiency causes salt-WASTING (hypotension, hyponatraemia) in severe forms — the opposite of hypertension. It is the 11β-hydroxylase and 17α-hydroxylase deficiencies that cause hypertension (due to DOC/corticosterone mineralocorticoid accumulation). 21-OH deficiency cannot complete aldosterone synthesis → salt wasting. 📝 How this gets tested: A CAH question with hypertension + virilisation = 11β-hydroxylase deficiency. A CAH question with salt-wasting + virilisation = 21-hydroxylase deficiency.
❌ Mistake: “In Addison disease, cortisol AND aldosterone are always both deficient” ✅ Reality: In PRIMARY adrenal insufficiency (Addison disease), BOTH cortisol AND aldosterone are deficient (because the entire adrenal cortex is destroyed — all three zones). In SECONDARY adrenal insufficiency (pituitary/hypothalamic failure), only cortisol is deficient — aldosterone production is largely ACTH-independent and remains intact. This is why secondary adrenal insufficiency does NOT cause hyperkalaemia or severe salt-wasting. 📝 How this gets tested: Hyperkalaemia + hyponatraemia + hyperpigmentation + low ACTH = does NOT exist. Primary Addison = ↑ ACTH. Secondary AI = ↓ ACTH but NO hyperkalaemia. Know which features distinguish them.
❌ Mistake: “All Cushing syndrome causes hyperpigmentation” ✅ Reality: Hyperpigmentation only occurs when ACTH is elevated (because ACTH and α-MSH both derive from POMC and cross-react with melanocortin receptors). Hyperpigmentation occurs in: Cushing disease (pituitary adenoma, elevated ACTH), ectopic ACTH syndrome (very elevated ACTH), and Addison disease (very elevated ACTH). It does NOT occur in: adrenal adenoma/carcinoma (low ACTH — the adrenal tumour produces cortisol autonomously, suppressing ACTH), or exogenous glucocorticoid use (exogenous GC suppresses ACTH). 📝 How this gets tested: Cushing syndrome + hyperpigmentation = ACTH-dependent cause. No hyperpigmentation + Cushing features + low ACTH = adrenal adenoma or exogenous GC.
❌ Mistake: “Short Synacthen test uses cortisol injection” ✅ Reality: The Short Synacthen test uses synthetic ACTH (tetracosactide, a 24-amino acid analogue of ACTH 1-24), NOT cortisol. It tests the ability of the adrenal to RESPOND to ACTH. A normal response (cortisol peak >550 nmol/L at 30 or 60 minutes) excludes primary adrenal insufficiency. It may give a false-normal in EARLY secondary adrenal insufficiency (adrenals not yet atrophied) — in which case, an insulin tolerance test or CRH test is needed. 📝 How this gets tested: “What is given in the Synacthen test?” = ACTH analogue (not cortisol). The point is to test adrenal reserve — if the adrenal fails to respond, it has insufficient functional cortex.
❌ Mistake: “Dexamethasone is used to replace cortisol in Addison disease” ✅ Reality: Dexamethasone has ZERO mineralocorticoid activity and a very long duration of action — it is not suitable for physiological cortisol replacement. Hydrocortisone is used for cortisol replacement (it has some mineralocorticoid activity and a short half-life matching physiological rhythm). Dexamethasone is used for anti-inflammatory/pharmacological purposes, DST, and specific clinical scenarios (cerebral oedema, foetal lung maturation). 📝 How this gets tested: “Which steroid is used for replacement in Addison disease?” = Hydrocortisone + Fludrocortisone. Dexamethasone is the wrong answer for replacement.
📝 5 Practice MCQs — Test Yourself Now
Q1: Which enzyme catalyses the rate-limiting step of cortisol biosynthesis, and which protein facilitates the delivery of substrate to this enzyme?
- A. CYP21A2 (21-hydroxylase); mediated by ACTH receptor
- B. CYP11A1 (P450scc); facilitated by StAR protein
- C. CYP17A1 (17α-hydroxylase); facilitated by CBG
- D. 3β-HSD; facilitated by ACTH
<details> <summary>👉 Click to reveal answer</summary>
✅ Answer: B. CYP11A1 (P450scc); facilitated by StAR protein
Why correct: CYP11A1 (cholesterol side-chain cleavage enzyme, P450scc) catalyses the rate-limiting step of ALL steroid hormone synthesis: Cholesterol → Pregnenolone. This enzyme is located on the inner mitochondrial membrane, and since cholesterol is stored in the cytoplasm/ER, it must be transported to the inner mitochondrial membrane. This transport is facilitated by StAR (Steroidogenic Acute Regulatory protein), which is the acute regulatory protein. ACTH stimulates StAR protein expression and phosphorylation — this is the primary mechanism by which ACTH acutely increases cortisol output within minutes.
Why A is wrong: CYP21A2 (21-hydroxylase) catalyses progesterone → 11-deoxycorticosterone and 17-OHP → 11-deoxycortisol — important steps but not the rate-limiting step. The ACTH receptor (MC2R) initiates the signalling cascade but does not catalyse or facilitate steroid synthesis directly. Why C is wrong: CYP17A1 (17α-hydroxylase) is an important step specific to cortisol and sex steroid synthesis (absent in ZG). CBG (cortisol-binding globulin) is a transport protein in blood, not involved in adrenal steroidogenesis. Why D is wrong: 3β-HSD catalyses Pregnenolone → Progesterone (Step 2). ACTH does stimulate steroidogenesis overall, but the specific facilitator of substrate delivery to CYP11A1 is StAR.
Exam tip: “Rate-limiting enzyme of steroid synthesis” = P450scc/CYP11A1. “Acute regulator of cortisol output” = StAR protein. Both are direct exam targets. ACTH stimulates both StAR and CYP11A1 — this dual action allows both acute and sustained increases in steroidogenesis. </details>
Q2: A newborn female presents at day 7 with vomiting, poor feeding, and shock. Blood results show Na⁺ 118 mmol/L, K⁺ 7.8 mmol/L, glucose 2.1 mmol/L. External genitalia appear ambiguous (clitoromegaly). 17-Hydroxyprogesterone is markedly elevated. Which enzyme is deficient and what is the treatment?
- A. 11β-Hydroxylase; treated with fludrocortisone only
- B. 17α-Hydroxylase; treated with hydrocortisone only
- C. 21-Hydroxylase; treated with hydrocortisone + fludrocortisone
- D. 3β-Hydroxysteroid dehydrogenase; treated with dexamethasone
✅ Answer: C. 21-Hydroxylase; treated with hydrocortisone + fludrocortisone
Why correct: This is the classic presentation of classic salt-wasting 21-hydroxylase (CYP21A2) deficiency CAH. The key clues: neonatal salt-wasting crisis (hyponatraemia + hyperkalaemia + shock), female virilisation (ambiguous genitalia in 46,XX), hypoglycaemia (cortisol deficiency → impaired gluconeogenesis), and the diagnostic hallmark of markedly elevated 17-OHP (the substrate that accumulates when 21-OH is absent). Treatment requires both hydrocortisone (replaces cortisol + suppresses ACTH → reduces androgen excess) and fludrocortisone (replaces aldosterone → corrects salt-wasting).
Why A is wrong: 11β-Hydroxylase deficiency causes virilisation but with HYPERTENSION (from DOC accumulation), not salt-wasting. 17-OHP would not be specifically elevated; 11-deoxycortisol and DOC would accumulate instead. Also, fludrocortisone alone would be insufficient and inappropriate. Why B is wrong: 17α-Hydroxylase deficiency causes inability to make sex hormones → no virilisation (the infant would have female phenotype even if 46,XY). It causes HTN and hypogonadism, not salt-wasting virilisation. Females would have primary amenorrhoea, not ambiguous genitalia. Why D is wrong: 3β-HSD deficiency impairs all steroid synthesis and can cause salt-wasting, but the specific marker for 21-OH deficiency is elevated 17-OHP. In 3β-HSD deficiency, it is 17-hydroxypregnenolone and DHEA that accumulate, not 17-OHP specifically. Also, dexamethasone is not the appropriate steroid for CAH replacement (too long-acting, no mineralocorticoid activity).
Exam tip: 17-OHP elevation is the specific diagnostic marker of 21-OH deficiency CAH. Salt-wasting + virilisation in a female neonate = 21-OH deficiency until proven otherwise. Hydrocortisone + fludrocortisone = complete replacement for classic CAH.
Q3: A 45-year-old man with known adrenal insufficiency on replacement hydrocortisone is admitted with appendicitis requiring emergency surgery. The surgical team is considering general anaesthesia. What is the most important perioperative management consideration?
- A. Stop hydrocortisone 24 hours pre-operatively to avoid immunosuppression
- B. Give supplemental hydrocortisone stress dosing — 100 mg IV at induction and every 6–8 hours post-op
- C. Switch to dexamethasone for better anti-inflammatory cover during surgery
- D. No adjustment needed — regular oral hydrocortisone can be continued as normal
✅ Answer: B. Give supplemental hydrocortisone stress dosing — 100 mg IV at induction and every 6–8 hours post-op
Why correct: Patients with adrenal insufficiency on replacement therapy cannot mount a normal cortisol stress response to surgery. Normally, a healthy person’s cortisol rises 5–10× baseline during major surgery to maintain haemodynamic stability, glucose, and vasopressor responsiveness. An adrenal-insufficient patient on standard replacement doses (15–25 mg/day) cannot do this. Without stress dosing, they risk life-threatening adrenal crisis (cardiovascular collapse, refractory hypotension) under anaesthesia. The standard protocol for major surgery is hydrocortisone 100 mg IV at induction followed by 50–100 mg every 6–8 hours for 24–48 hours, then taper back to maintenance once they can tolerate oral intake.
Why A is wrong: Stopping hydrocortisone is dangerous and could precipitate adrenal crisis. Patients with adrenal insufficiency need their baseline replacement PLUS additional stress dosing — NOT removal of their regular dose. Why C is wrong: Dexamethasone has no mineralocorticoid activity and does not provide adequate physiological cortisol replacement. It is not appropriate for acute stress dosing in adrenal insufficiency where both glucocorticoid and mineralocorticoid-like effects are needed. Additionally, dexamethasone would interfere with cortisol monitoring. Why D is wrong: Oral absorption may be unreliable perioperatively (fasting, ileus, haemodynamic instability). Regular maintenance doses (15–25 mg/day) are grossly insufficient for major surgical stress — surgical stress typically requires the equivalent of 200–300 mg cortisol/day in a normal person’s response.
Exam tip: “Sick day rules” for adrenal insufficiency = double or triple the oral steroid dose for illness/minor procedures, and give IV hydrocortisone for major surgery/inability to take orally. This is one of the most clinically important management facts about cortisol physiology — an iatrogenic omission of stress dosing can kill.
Q4: A 28-year-old woman on the oral contraceptive pill has a random serum cortisol of 780 nmol/L (elevated; normal 170–690 nmol/L in the afternoon). She is completely well with no features of Cushing syndrome. What is the most likely explanation?
- A. Early Cushing syndrome — refer for formal DST
- B. OCP increases CBG synthesis → increased total serum cortisol, but free cortisol is normal
- C. The OCP is directly stimulating cortisol secretion from the adrenal gland
- D. The OCP is inhibiting 11β-HSD2, causing cortisol accumulation
✅ Answer: B. OCP increases CBG synthesis → increased total serum cortisol, but free cortisol is normal
Why correct: Oestrogen (in the OCP and in pregnancy) stimulates hepatic synthesis of cortisol-binding globulin (CBG/transcortin). CBG binds cortisol with high affinity. When CBG rises, more cortisol is bound → less is excreted or metabolised → total serum cortisol rises. However, the FREE cortisol fraction (the biologically active form) remains normal because cortisol secretion from the adrenal is unchanged (the HPA axis is intact and regulates free cortisol). Since this woman has no clinical features of Cushing syndrome, and the biochemical change is purely due to elevated CBG, no further investigation is needed. This is confirmed by measuring urinary free cortisol (UFC) or late-night salivary cortisol — both should be normal.
Why A is wrong: Referral for DST is unnecessary in an asymptomatic woman on OCP with elevated total cortisol. Performing a DST in this context would be wasteful — the DST measures total cortisol suppression, which would still appear blunted because of elevated CBG. The appropriate next step would be checking UFC or stopping OCP and rechecking, not DST. Why C is wrong: OCP does not directly stimulate adrenal cortisol secretion. The adrenal is regulated by ACTH via the HPA axis. OCP effects on cortisol are mediated entirely through increased CBG production in the liver. Why D is wrong: 11β-HSD2 converts cortisol → cortisone in the kidney. Inhibition (as in liquorice ingestion) would cause apparent mineralocorticoid excess (hypertension, hypokalaemia) — not elevated serum cortisol levels. The OCP does not inhibit 11β-HSD2.
Exam tip: OCP / pregnancy + elevated serum cortisol + NO clinical Cushing features = elevated CBG — not real hypercortisolism. Free cortisol (UFC or salivary) is NORMAL. This is a classic confounding scenario in Cushing syndrome workup. Always correlate total cortisol with clinical features and CBG status.
Q5: A 60-year-old man with small cell lung carcinoma (SCLC) develops profound hypokalaemia (K⁺ 2.0 mmol/L), hypertension, oedema, and severe proximal myopathy. Serum cortisol is 2,800 nmol/L (extremely elevated) and ACTH is 950 pg/mL (markedly elevated). A low-dose DST shows no suppression; a high-dose DST also shows no suppression. What is the diagnosis and why is hyperpigmentation likely present?
- A. Cushing disease — pituitary adenoma with ectopic ACTH-like molecule
- B. Ectopic ACTH syndrome — autonomous ACTH from SCLC; hyperpigmentation from extreme ACTH → α-MSH excess
- C. Adrenal carcinoma — autonomous cortisol production; hyperpigmentation from direct melanocyte stimulation by cortisol
- D. Primary adrenal hyperplasia — bilateral nodular disease; low ACTH causing hyperpigmentation via unknown mechanism
✅ Answer: B. Ectopic ACTH syndrome — autonomous ACTH from SCLC; hyperpigmentation from extreme ACTH → α-MSH excess
Why correct: This is the classic presentation of ectopic ACTH syndrome. SCLC (and other neuroendocrine tumours) can autonomously secrete ACTH. The extremely elevated ACTH and cortisol, failure to suppress on BOTH low and high-dose DST (autonomous secretion unresponsive to glucocorticoid feedback), and the clinical syndrome (hypokalaemia is particularly severe in ectopic ACTH because extremely high cortisol levels overwhelm 11β-HSD2 in the kidney → cortisol binds mineralocorticoid receptors → massive Na⁺ retention and K⁺ loss) all point to ectopic ACTH. Hyperpigmentation occurs because ACTH and α-MSH are both cleaved from the same precursor POMC — when ACTH is massively elevated, so is α-MSH, which stimulates melanocytes. The rapid onset and severity (metabolic derangement > physical signs in many ectopic cases) are characteristic.
Why A is wrong: Cushing disease (pituitary adenoma) has elevated ACTH but typically SUPPRESSES on high-dose DST (pituitary adenoma cells still retain some GR responsiveness and respond to high GC doses). Failure to suppress on high-dose DST excludes a pituitary source. Why C is wrong: Adrenal carcinoma produces cortisol autonomously → suppresses ACTH (ACTH would be undetectable, not 950 pg/mL). Cortisol itself does not cause hyperpigmentation — it is ACTH (via α-MSH) that does. With an adrenal source, ACTH is low → NO hyperpigmentation. Why D is wrong: Bilateral adrenal hyperplasia is a consequence of ACTH excess, not an autonomous primary process. ACTH would not be low in primary hyperplasia. Low ACTH never causes hyperpigmentation — hyperpigmentation specifically requires ACTH elevation.
Exam tip: Ectopic ACTH syndrome: Very high ACTH + cortisol + no DST suppression (low OR high dose) + severe hypokalaemia (overwhelming MR) + rapid onset + underlying malignancy (SCLC most common). IPSS sampling shows no central:peripheral gradient (confirms extra-pituitary source).
📚 References
📖 Harper’s Illustrated Biochemistry — Rodwell et al. | Chapter 40: The Adrenocortical Steroids; Chapter 26: Hormones of the Adrenal Cortex
📖 Lippincott’s Illustrated Reviews: Biochemistry — Ferrier | Chapter 23: Steroid Hormone Synthesis
📖 Guyton and Hall Textbook of Medical Physiology — Hall | Chapter 77: Adrenocortical Hormones
📖 Williams Textbook of Endocrinology — Melmed et al. | Chapter 15: The Adrenal Cortex
📖 Greenspan’s Basic & Clinical Endocrinology — Gardner & Shoback | Chapter 9: Glucocorticoids and Adrenal Androgens
📖 Katzung’s Basic & Clinical Pharmacology — Katzung | Chapter 39: Adrenocorticosteroids and Adrenocortical Antagonists
🚀 Keep Practising — You Are Not Done Yet
Cortisol questions in NEET PG and USMLE are almost always clinical scenarios — a neonate with ambiguous genitalia, a woman with resistant hypertension on OCP, a man with SCLC and hypokalaemia, a post-surgical patient in shock. The biochemistry is the decoder that explains why each sign and symptom occurs.
The students who score on cortisol questions are the ones who understand the steroidogenesis pathway well enough to predict what accumulates when each enzyme fails — and who understand the HPA axis well enough to know which tests distinguish the causes.
medicalmcq.in has free MCQs covering every aspect of cortisol — steroidogenesis, HPA axis, Cushing syndrome, CAH, Addison disease, and glucocorticoid pharmacology — all in clinical-scenario format with detailed mechanistic explanations.