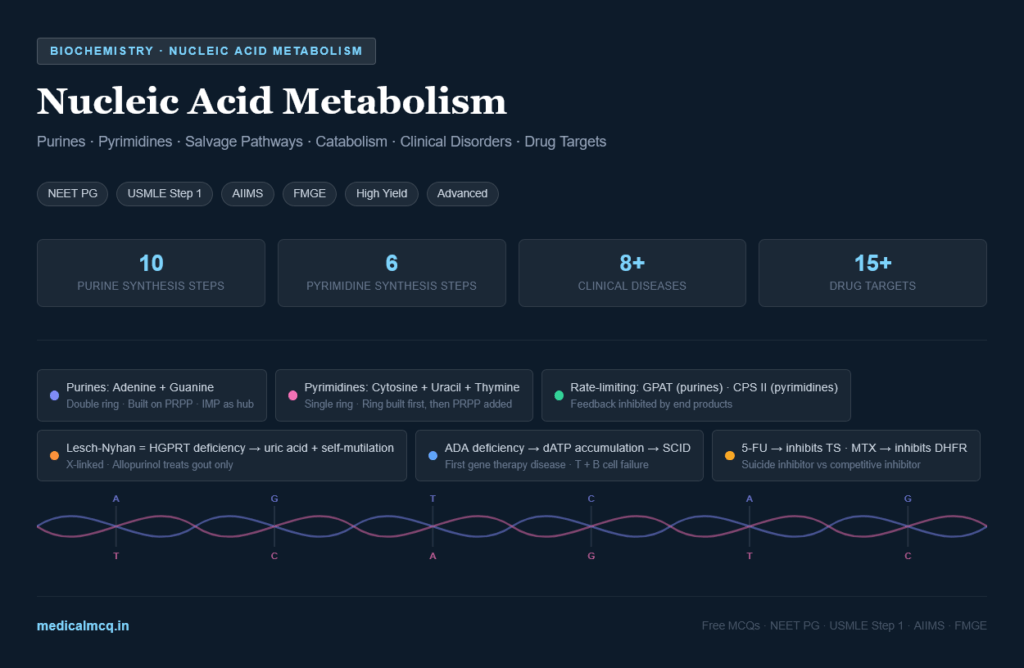
What You Will Learn in This Article
- Distinguish purines from pyrimidines by structure and name every base, nucleoside, and nucleotide
- Trace the de novo synthesis of purines step by step — all 10 steps with enzymes and substrates
- Trace the de novo synthesis of pyrimidines — all 6 steps with the unique features of each
- Explain the salvage pathways and why their deficiency causes distinct diseases
- State the complete catabolism pathways for purines and pyrimidines including the final products
- Identify the rate-limiting enzymes and their regulators for both synthesis pathways
- Connect nucleotide metabolism to clinical disorders: gout, HGPRT deficiency (Lesch-Nyhan), adenosine deaminase deficiency (SCID), orotic aciduria
- Explain the mechanisms of anti-cancer and anti-viral drugs that target nucleotide metabolism
📖 Introduction: Why This Topic Matters in Exams
A 3-year-old boy is brought in for self-mutilation — he bites his own fingers and lips repeatedly, and the parents report he cannot stop himself. He has intellectual disability, choreoathetosis, and spastic cerebral palsy. Uric acid in blood and urine is massively elevated. The diagnosis is Lesch-Nyhan syndrome — a deficiency of HGPRT (hypoxanthine-guanine phosphoribosyltransferase), the key enzyme of the purine salvage pathway. Without this enzyme, hypoxanthine and guanine cannot be recycled back into usable nucleotides and instead flood the xanthine oxidase pathway, generating enormous quantities of uric acid. The compulsive self-mutilation is one of the most specific clinical signs in all of biochemistry.
Nucleic acid metabolism is tested intensively across NEET PG, USMLE Step 1, and AIIMS. Questions range from pure recall (“what is the rate-limiting enzyme of purine synthesis?”) to drug mechanism questions (“why does methotrexate cause megaloblastic anaemia?”) to full clinical scenarios (“a child with recurrent infections and lymphopenia — which enzyme is deficient?”). The pharmacology of nucleotide metabolism — anticancer drugs, antivirals, gout medications — is among the most clinically relevant biochemistry in the entire curriculum.
This article covers the complete landscape: purine and pyrimidine structure, de novo synthesis, salvage pathways, catabolism, clinical disorders, and drug mechanisms. Work through it systematically — the pathway logic is the key, not isolated facts.
🔬 Section 1 — Structure: Purines vs Pyrimidines
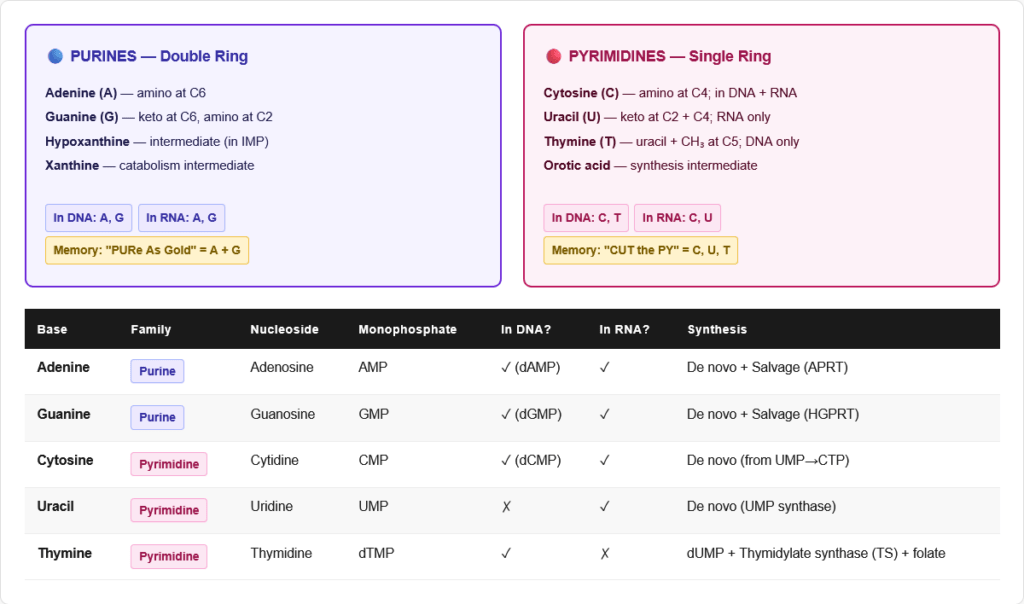
The Two Families
Nucleic acid bases are divided into two structural families:
Purines — double-ring structure (fused pyrimidine + imidazole rings)
- Adenine (A) — amino group at C6
- Guanine (G) — keto group at C6, amino group at C2
Pyrimidines — single-ring structure (6-membered ring only)
- Cytosine (C) — amino group at C4
- Uracil (U) — found in RNA only; two keto groups
- Thymine (T) — found in DNA only; uracil + methyl group at C5
Memory rule: “CUT the PY” — Cytosine, Uracil, Thymine are PYrimidines. Adenine and Guanine are PURines — “PURe As Gold.”
Nucleosides and Nucleotides
| Level | Definition | Example |
|---|---|---|
| Base | The nitrogenous ring compound alone | Adenine |
| Nucleoside | Base + sugar (ribose or deoxyribose) | Adenosine (AMP without phosphate) |
| Nucleotide | Nucleoside + 1–3 phosphate groups | AMP, ADP, ATP |
Sugar used:
- RNA nucleotides: ribose (2′-OH present)
- DNA nucleotides: deoxyribose (2′-H, no OH)
Purine Nucleotides
| Base | Nucleoside | Monophosphate | Diphosphate | Triphosphate |
|---|---|---|---|---|
| Adenine | Adenosine | AMP | ADP | ATP |
| Guanine | Guanosine | GMP | GDP | GTP |
| Hypoxanthine | Inosine | IMP | — | — |
| Xanthine | Xanthosine | XMP | — | — |
Pyrimidine Nucleotides
| Base | Nucleoside | Monophosphate | Notes |
|---|---|---|---|
| Cytosine | Cytidine | CMP | In RNA; DNA (as dCMP) |
| Uracil | Uridine | UMP | RNA only |
| Thymine | Thymidine | TMP (dTMP) | DNA only; made from dUMP |
| Orotic acid | Orotidine | OMP | Intermediate in synthesis |
🔬 Section 2 — De Novo Purine Synthesis: Building the Ring Atom by Atom
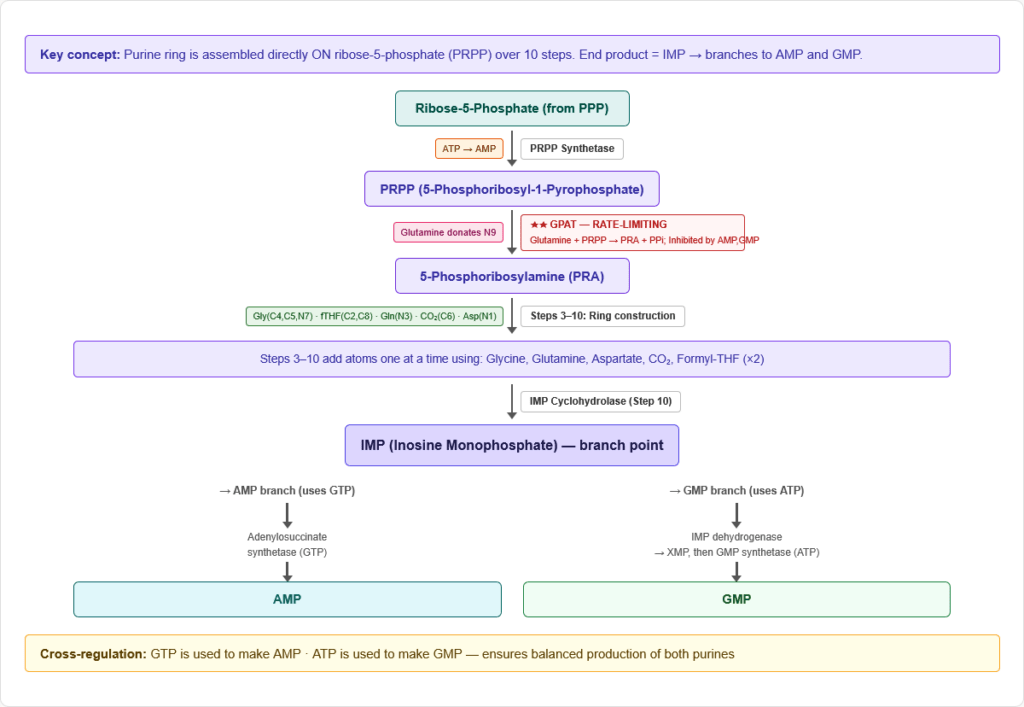
The Concept
De novo purine synthesis builds the purine ring directly on ribose-5-phosphate (not on a preformed base). The ring is constructed one atom at a time over 10 steps, starting from phosphoribosyl pyrophosphate (PRPP) and ending at inosine monophosphate (IMP), the common precursor to both AMP and GMP.
Location: Cytoplasm (all steps) Starting material: Ribose-5-phosphate (from pentose phosphate pathway) End product: IMP → AMP and GMP
Atom Origins in the Purine Ring
This is one of the most tested facts about purine synthesis:
| Atom in purine ring | Donor |
|---|---|
| N1 | Aspartate |
| C2 | Formyl-THF (one-carbon, folate) |
| N3 | Glutamine |
| C4 | Glycine |
| C5 | Glycine |
| C6 | CO₂ |
| N7 | Glycine |
| C8 | Formyl-THF (one-carbon, folate) |
| N9 | Glutamine |
Quick summary: “Glycine makes the scaffold (C4, C5, N7), Glutamine donates 2 nitrogens (N3, N9), Aspartate gives N1, CO₂ gives C6, and Folate (as formyl-THF) gives C2 and C8.”
The 10 Steps of De Novo Purine Synthesis
Step 1 — PRPP synthesis (prerequisite step)
- Ribose-5-phosphate + ATP → PRPP (5-phosphoribosyl-1-pyrophosphate) + AMP
- Enzyme: PRPP synthetase (PRPP synthase)
- PRPP is the activated ribose that all nucleotide synthesis starts from
- PRPP synthetase is activated by inorganic phosphate and inhibited by purine nucleotides (feedback)
Step 2 — Committed step: PRPP → PRA
- PRPP + Glutamine → 5-phosphoribosylamine (PRA) + Glutamate + PPi
- Enzyme: Glutamine phosphoribosylpyrophosphate amidotransferase (GPAT) — rate-limiting enzyme of de novo purine synthesis
- This is the irreversible, committed step — the point of no return
- Inhibited by: AMP, ADP, ATP, GMP, GDP, GTP (all end products — feedback inhibition)
- Activated by: PRPP
Steps 3–10 — Ring construction
| Step | Addition | Enzyme | Cofactor |
|---|---|---|---|
| 3 | Glycine added (C4, C5, N7) | GAR synthetase | ATP |
| 4 | Formyl group from formyl-THF (C8) | GAR transformylase | Folate (N10-formyl-THF) |
| 5 | Amide from glutamine (N3) | FGAM synthetase | ATP, Glutamine |
| 6 | Ring closure → imidazole formed | AIR synthetase | ATP |
| 7 | CO₂ carboxylation (C6) | CAIR synthetase | ATP, CO₂ |
| 8 | Aspartate addition (N1) | SAICAR synthetase | ATP, Aspartate |
| 9 | Fumarate release, N1 remains | Adenylosuccinate lyase | — |
| 10 | Second formyl group from formyl-THF (C2) | AICAR transformylase | Folate (N10-formyl-THF) |
| Final | Ring closure → IMP formed | IMP cyclohydrolase | — |
End product: IMP (Inosine Monophosphate)
IMP → AMP and GMP (Branching from IMP)
IMP → AMP (two steps):
- IMP + Aspartate → Adenylosuccinate (enzyme: Adenylosuccinate synthetase; uses GTP as energy source)
- Adenylosuccinate → AMP + Fumarate (enzyme: Adenylosuccinate lyase)
IMP → GMP (two steps):
- IMP → XMP (enzyme: IMP dehydrogenase; uses NAD⁺)
- XMP + Glutamine → GMP (enzyme: GMP synthetase; uses ATP)
Cross-regulation:
- GTP is used to make AMP; ATP is used to make GMP
- This ensures balanced production of both purines — if GTP is high, it drives AMP synthesis; if ATP is high, it drives GMP synthesis
Regulation of De Novo Purine Synthesis
| Regulator | Effect | Mechanism |
|---|---|---|
| AMP, ADP, ATP | Inhibit | Feedback inhibition of GPAT (committed step) and PRPP synthetase |
| GMP, GDP, GTP | Inhibit | Same — feedback inhibition |
| PRPP (high) | Activates | Substrate activates GPAT |
| AMP inhibits AMP synthesis | Further inhibition | AMP inhibits adenylosuccinate synthetase (IMP→AMP branch) |
| GMP inhibits GMP synthesis | Further inhibition | GMP inhibits IMP dehydrogenase (IMP→GMP branch) |
🔬 Section 3 — De Novo Pyrimidine Synthesis
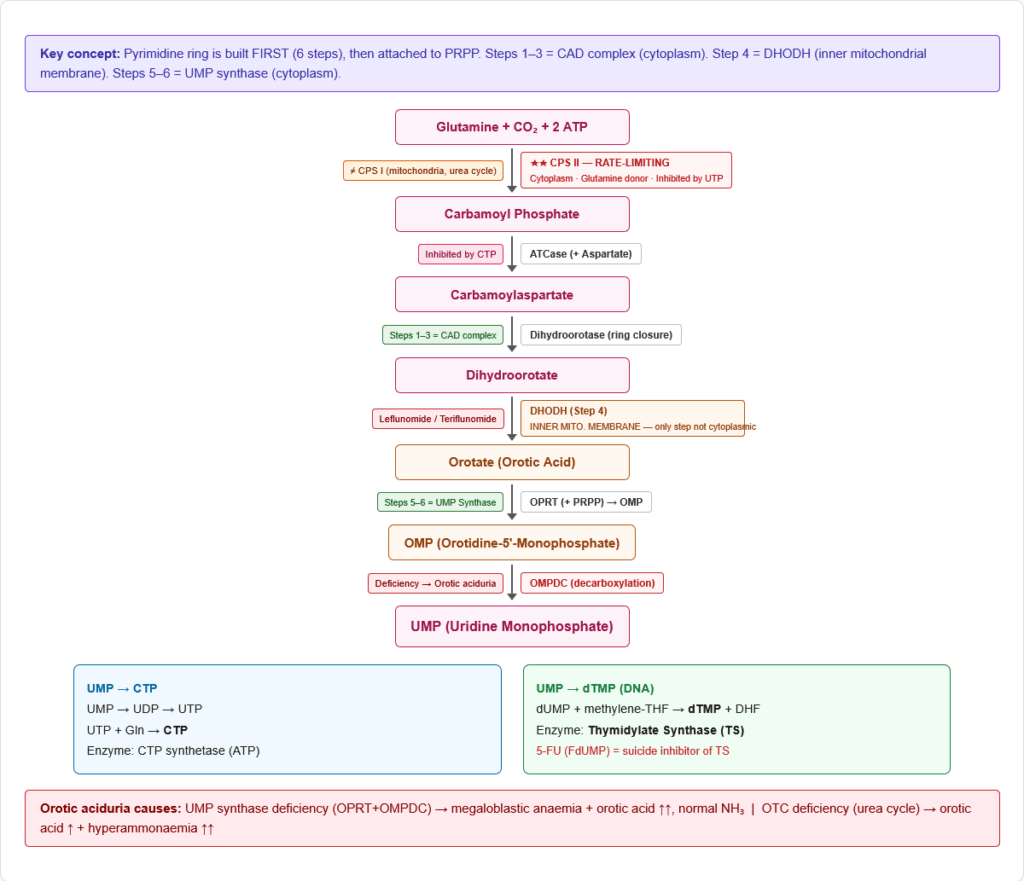
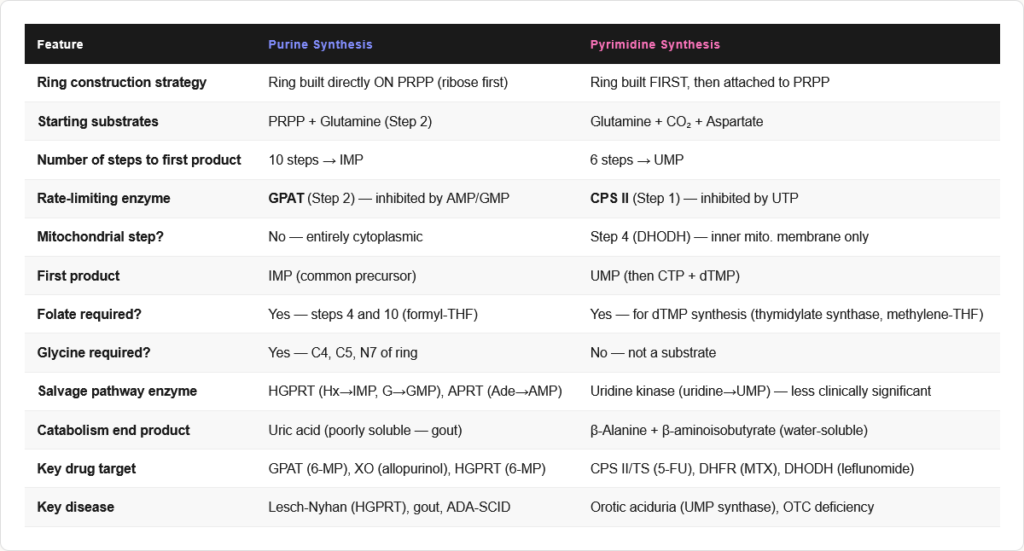
Key Differences from Purine Synthesis
| Feature | Purine Synthesis | Pyrimidine Synthesis |
|---|---|---|
| Ring built on | Ribose-5-P (PRPP) | Ring built FIRST, then attached to PRPP |
| Starting material | PRPP | Carbamoyl phosphate + Aspartate |
| End product (first) | IMP | UMP |
| Rate-limiting enzyme | GPAT (Step 2) | CAD complex / Carbamoyl phosphate synthetase II |
| Compartment | Cytoplasm | Cytoplasm (steps 1–3 by CAD) + mitochondria (CPS I for urea cycle, NOT pyrimidines) |
The 6 Steps of Pyrimidine Synthesis
Step 1 — Carbamoyl phosphate synthesis
- Glutamine + CO₂ + 2 ATP → Carbamoyl phosphate
- Enzyme: Carbamoyl phosphate synthetase II (CPS II) — cytoplasmic
- Rate-limiting enzyme of pyrimidine synthesis
- Inhibited by: UTP (feedback)
- Activated by: PRPP, ATP
- Note: CPS I (mitochondrial, uses ammonia) is for the urea cycle. CPS II (cytoplasmic, uses glutamine) is for pyrimidines. These are completely different enzymes at different locations with different substrates.
Step 2 — Formation of carbamoylaspartate
- Carbamoyl phosphate + Aspartate → Carbamoylaspartate
- Enzyme: Aspartate transcarbamoylase (ATCase)
- The classic allosteric enzyme used in biochemistry teaching: inhibited by CTP (end product), activated by ATP
Step 3 — Ring closure
- Carbamoylaspartate → Dihydroorotate
- Enzyme: Dihydroorotase
Steps 1–3 in mammals are all performed by a single trifunctional enzyme: CAD (Carbamoyl phosphate synthetase II / Aspartate transcarbamoylase / Dihydroorotase)
Step 4 — Oxidation to orotate
- Dihydroorotate → Orotate
- Enzyme: Dihydroorotate dehydrogenase (DHODH)
- Location: Inner mitochondrial membrane — the ONLY pyrimidine synthesis step in mitochondria
- Inhibited by: Leflunomide/Teriflunomide (active metabolite A771726) — used in rheumatoid arthritis and multiple sclerosis
Step 5 — Attachment to PRPP
- Orotate + PRPP → Orotidine-5′-monophosphate (OMP)
- Enzyme: Orotate phosphoribosyltransferase (OPRT)
- OMP contains the pyrimidine ring attached to ribose
Step 6 — Decarboxylation → UMP
- OMP → UMP (Uridine monophosphate)
- Enzyme: OMP decarboxylase (OMPDC)
- Inhibited by: UMP, CMP (product feedback)
Steps 5 and 6 are carried out by a bifunctional enzyme: UMP synthase (OPRT + OMPDC)
Deficiency of UMP synthase → Hereditary Orotic Aciduria:
- Orotic acid accumulates (cannot be converted to OMP or UMP) → excreted in urine (orotic aciduria)
- Megaloblastic anaemia (cannot make pyrimidines for DNA synthesis) — does NOT respond to B12 or folate
- Treatment: Uridine supplementation (bypasses the block — UMP can be made from uridine by uridine kinase)
UMP → CTP and dTMP
UMP → CTP:
- UMP → UDP → UTP (kinases)
- UTP + Glutamine → CTP (enzyme: CTP synthetase; uses ATP)
UMP → dTMP (thymidylate synthesis — critical for DNA):
- UDP → dUDP → dUMP (ribonucleotide reductase converts ribose to deoxyribose)
- dUMP + N5,N10-methylene-THF → dTMP + DHF
- Enzyme: Thymidylate synthase (TS)
- DHF → THF (dihydrofolate reductase — DHFR; requires NADPH)
- This reaction is the only source of thymidine for DNA synthesis
Drug targets at this step:
- 5-Fluorouracil (5-FU): Converted to FdUMP → irreversibly inhibits thymidylate synthase (suicide inhibitor) → no dTMP → DNA synthesis blocked → cancer cells die
- Methotrexate (MTX): Inhibits DHFR → DHF cannot be converted back to THF → thymidylate synthase reaction stalls (no THF to donate methylene group) → dTMP production stops + folate-dependent purine synthesis also inhibited → megaloblastic changes
- Hydroxyurea: Inhibits ribonucleotide reductase (RNR) → blocks dNTP synthesis
⚙️ Section 4 — Purine Salvage Pathways
Why Salvage Matters
De novo synthesis of nucleotides is energetically expensive (6 ATP per purine ring). The salvage pathway recycles free bases (from nucleotide degradation) back into usable nucleotides using a single phosphoribosyltransferase reaction:
Base + PRPP → Nucleotide + PPi
This is far cheaper than de novo synthesis and accounts for the majority of nucleotide recycling in tissues.
The Two Key Salvage Enzymes
HGPRT (Hypoxanthine-Guanine Phosphoribosyltransferase):
- Recycles Hypoxanthine → IMP
- Recycles Guanine → GMP
- Location: Cytoplasm; present in all tissues; highest activity in brain
APRT (Adenine Phosphoribosyltransferase):
- Recycles Adenine → AMP
- Less clinically significant than HGPRT
- APRT deficiency → 2,8-dihydroxyadenine kidney stones (rare)
Lesch-Nyhan Syndrome (HGPRT Deficiency)
Genetics: X-linked recessive — affects males almost exclusively
Biochemistry:
- HGPRT absent → hypoxanthine and guanine cannot be salvaged → they accumulate
- Both hypoxanthine and guanine → degraded by xanthine oxidase → uric acid (massive overproduction)
- Loss of salvage also means loss of negative feedback on PRPP synthetase → de novo synthesis runs uncontrolled → more PRPP → more purines → more uric acid
- Combined effect: hyperuricaemia of extraordinary degree (often >10 mg/dL)
Clinical presentation:
- Severe hyperuricaemia → gout, urate nephropathy, uric acid stones
- Compulsive self-mutilation (lip/finger biting) — pathognomonic, neurological basis not fully understood
- Intellectual disability (moderate to severe)
- Choreoathetosis, dystonia, spastic cerebral palsy
- Orange/sandy urine (uric acid crystals in nappies)
Treatment:
- Allopurinol (xanthine oxidase inhibitor) → reduces uric acid production but does NOT fix the neurological manifestations
- No cure; neurological symptoms persist regardless of uric acid control
Adenosine Deaminase (ADA) Deficiency
- ADA converts Adenosine → Inosine and dAdo → dInosine
- ADA deficiency → dATP and dAdo accumulate in lymphocytes
- dATP: inhibits ribonucleotide reductase → blocks DNA synthesis in all cells; also directly toxic to lymphocytes
- Result: Severe Combined Immunodeficiency (SCID) — both T cells and B cells are decimated
- This is the “bubble boy” disease — affected children lack all adaptive immunity
- First disease treated by gene therapy (1990)
- Treatment: PEG-ADA (polyethylene glycol-modified ADA enzyme), bone marrow/stem cell transplant, gene therapy
⚙️ Section 5 — Purine Catabolism: From Nucleotides to Uric Acid
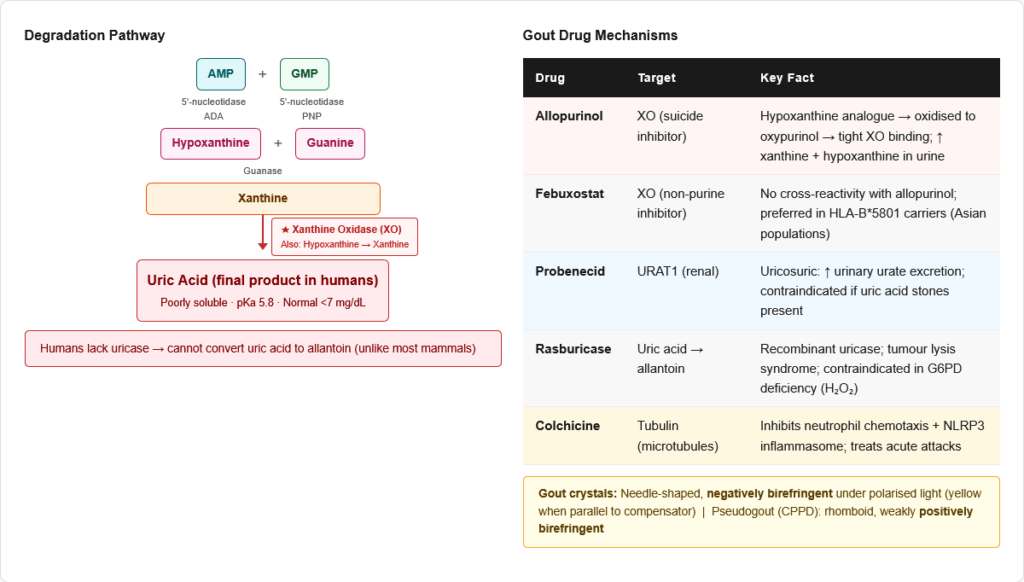
The Degradation Pathway
All purines are ultimately degraded to uric acid in humans (unlike most other mammals, which can convert uric acid to allantoin via uricase — humans lack this enzyme).
AMP degradation:
- AMP → Adenosine → Inosine → Hypoxanthine
- Enzymes: AMP deaminase / adenosine deaminase / purine nucleoside phosphorylase (PNP)
GMP degradation:
- GMP → Guanosine → Guanine → Xanthine
- Enzymes: 5′-nucleotidase / purine nucleoside phosphorylase / guanase
Final steps (both pathways converge):
- Hypoxanthine → Xanthine (enzyme: Xanthine oxidase (XO))
- Xanthine → Uric Acid (enzyme: Xanthine oxidase (XO))
- Xanthine oxidase is the key enzyme — it catalyses both final steps; it uses O₂ and generates H₂O₂ and superoxide (reactive oxygen species)
Uric acid:
- pKa = 5.8; mostly present as urate at physiological pH
- Poorly soluble, especially at low pH and temperature
- Normal plasma urate: <7 mg/dL (men), <6 mg/dL (women)
- Hyperuricaemia (>7 mg/dL) can lead to gout when crystals deposit in joints and soft tissues
Purine Nucleoside Phosphorylase (PNP) Deficiency
- PNP converts Inosine → Hypoxanthine and Guanosine → Guanine
- PNP deficiency → dGTP and dATP accumulate (particularly in T cells)
- Result: Primarily T cell immunodeficiency (unlike ADA deficiency which affects both T and B cells)
- B cells are relatively spared (they can use alternative pathways)
- Clinical: Recurrent infections, autoimmune features, neurological symptoms
⚙️ Section 6 — Pyrimidine Catabolism
Pyrimidines are catabolised very differently from purines — they produce non-toxic, water-soluble end products (not uric acid).
Cytosine → Uracil (deamination by cytidine deaminase)
Uracil degradation:
- Uracil → Dihydrouracil → Ureidopropionate → β-Alanine + CO₂ + NH₃
Thymine degradation:
- Thymine → Dihydrothymine → Ureidoisobutyrate → β-Aminoisobutyrate + CO₂ + NH₃
Key fact: β-Aminoisobutyrate from thymine catabolism is excreted in urine and can serve as a marker of increased cell turnover (cancer, chemotherapy, radiation) or increased DNA degradation.
Dihydropyrimidine dehydrogenase (DPD) deficiency:
- Rate-limiting enzyme of pyrimidine catabolism
- Deficiency → 5-fluorouracil (5-FU) cannot be degraded → severe 5-FU toxicity with standard doses (mucositis, myelosuppression, neurotoxicity)
- DPD deficiency testing before 5-FU therapy is now recommended in many countries
- Also causes mild neurological symptoms (due to accumulation of uracil and thymine)
🏥 Section 7 — Gout: Hyperuricaemia and Crystal Deposition Disease
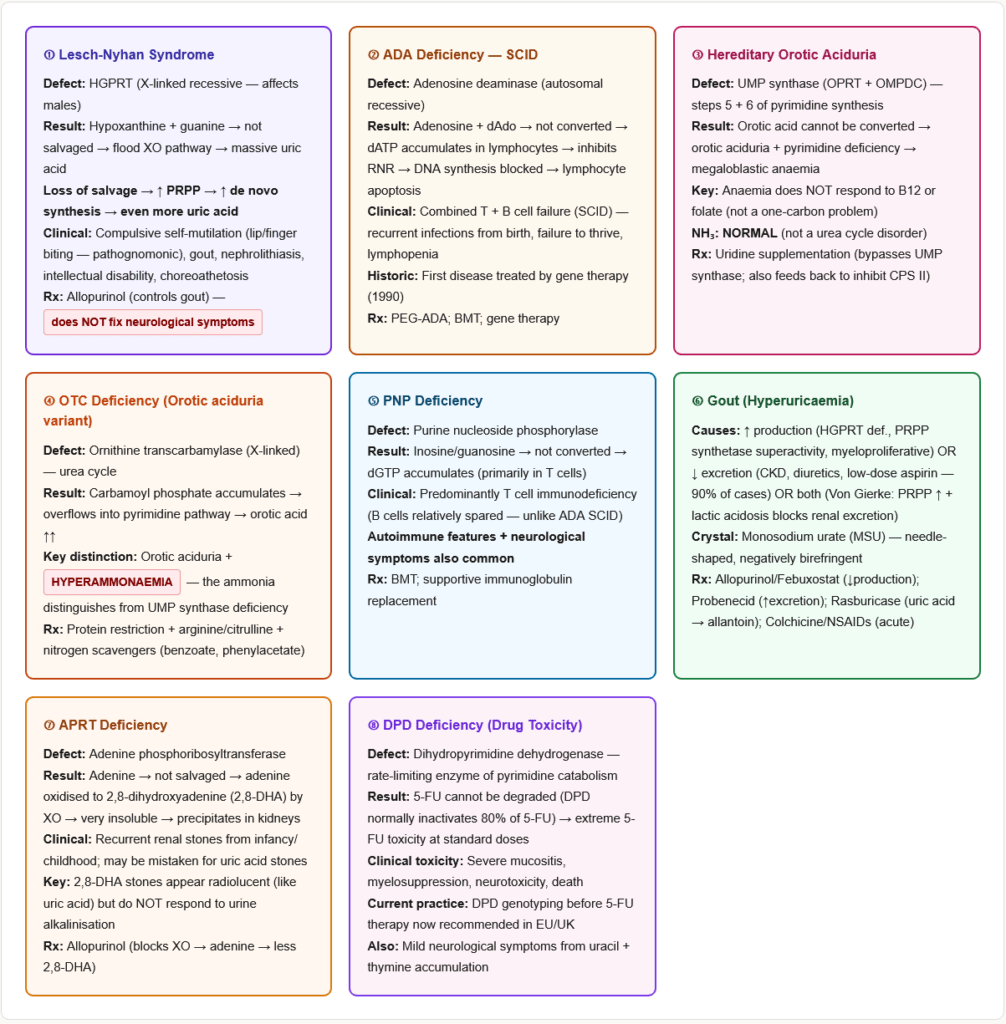
Pathophysiology
Gout results from chronic hyperuricaemia (urate >7 mg/dL) leading to monosodium urate (MSU) crystal deposition in joints and soft tissues.
Causes of hyperuricaemia:
- Overproduction (10%): HGPRT deficiency, PRPP synthetase superactivity, myeloproliferative disorders (increased cell turnover), haemolytic anaemia, psoriasis, high purine diet
- Underexcretion (90%): Chronic kidney disease (most common), diuretics (thiazides, furosemide — compete with urate for secretion), low-dose aspirin, alcohol, lead nephropathy (saturnine gout)
- Both: Glucose-6-phosphatase deficiency (Von Gierke disease) — excess G6P → pentose phosphate pathway → excess PRPP → purine overproduction + renal urate underexcretion from lactic acidosis
Crystal deposition: MSU crystals → activates NLRP3 inflammasome in macrophages → IL-1β and IL-18 release → acute inflammatory arthritis
Clinical stages:
- Asymptomatic hyperuricaemia
- Acute gouty arthritis (podagra — first MTP joint most common; also ankle, knee, wrist)
- Intercritical gout (symptom-free intervals)
- Chronic tophaceous gout (tophi in pinna, Achilles tendon, extensor surfaces)
Joint fluid: Negatively birefringent, needle-shaped crystals under polarised light (yellow when parallel to compensator)
Treatment of Gout
Acute attack:
- NSAIDs (indomethacin, naproxen — first line)
- Colchicine (inhibits microtubule polymerisation → impairs neutrophil chemotaxis and NLRP3 activation)
- Corticosteroids (if NSAIDs contraindicated)
Chronic/preventive:
- Allopurinol — structural analogue of hypoxanthine; competitive then suicide inhibitor of xanthine oxidase. Xanthine oxidase converts allopurinol → oxypurinol, which binds tightly (suicide inhibitor). Reduces uric acid production.
- Febuxostat — non-purine selective XO inhibitor; useful when allopurinol is contraindicated (allopurinol hypersensitivity, especially in HLA-B*5801 carriers — Han Chinese, Thai, Korean populations)
- Probenecid — inhibits urate reabsorption transporter (URAT1) in renal tubule → increases urate excretion (uricosuric agent)
- Rasburicase — recombinant uricase; converts uric acid → allantoin (soluble) → rapid urate lowering; used in tumour lysis syndrome. Contraindicated in G6PD deficiency (H₂O₂ generated damages RBCs)
- Pegloticase — pegylated uricase for refractory chronic gout
🔬 Section 8 — Drug Mechanisms Targeting Nucleotide Metabolism
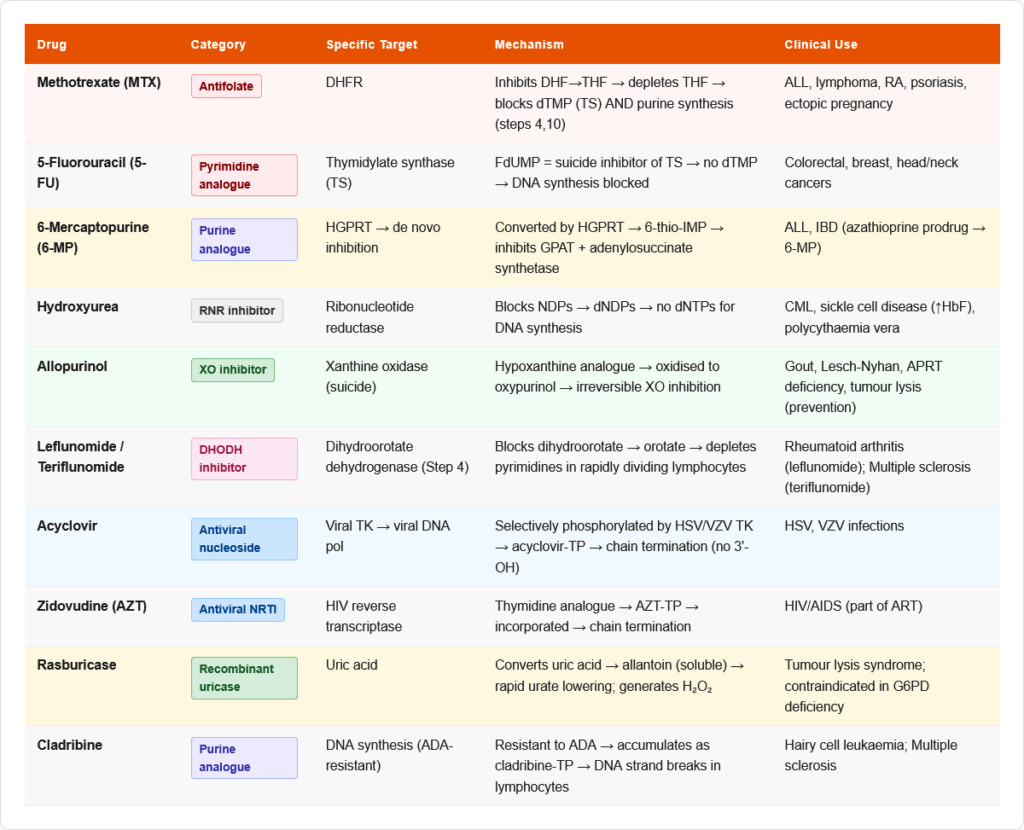
Anticancer Drugs
| Drug | Target | Mechanism | Clinical Use |
|---|---|---|---|
| Methotrexate (MTX) | DHFR | Inhibits DHF→THF conversion → depletes folate → blocks dTMP synthesis + purine synthesis → impairs DNA synthesis | Acute leukaemia, lymphoma, RA, psoriasis |
| 5-Fluorouracil (5-FU) | Thymidylate synthase | FdUMP (active metabolite) → irreversible “suicide” inhibitor of TS → no dTMP → DNA synthesis blocked | Colorectal, breast, head/neck cancers |
| 6-Mercaptopurine (6-MP) | HGPRT + multiple | Converted by HGPRT to 6-thio-IMP → inhibits de novo purine synthesis (blocks GPAT and adenylosuccinate synthetase); also incorporated into DNA | ALL (acute lymphoblastic leukaemia), IBD |
| 6-Thioguanine | Same as 6-MP | Similar mechanism; thio-GTP incorporated into DNA → strand breaks | ALL, AML |
| Hydroxyurea | Ribonucleotide reductase (RNR) | Inhibits RNR → blocks dNDP → dNTP conversion → no DNA synthesis | CML, sickle cell disease (↑ HbF), polycythaemia vera |
| Cytarabine (Ara-C) | DNA polymerase | Ara-CTP incorporated into DNA → chain termination | AML (acute myeloid leukaemia) |
| Gemcitabine | RNR + DNA polymerase | Difluorodeoxycytidine → inhibits RNR + incorporated → chain termination | Pancreatic, lung, bladder cancer |
| Pemetrexed | Multiple folate enzymes (TS, DHFR, GARFT) | Broader antifolate; requires polyglutamation for activity | Non-small cell lung cancer, mesothelioma |
| Cladribine | DNA synthesis in lymphocytes | Resistant to ADA → accumulates as cladribine triphosphate → DNA strand breaks, apoptosis | Hairy cell leukaemia, MS |
Antiviral Drugs
| Drug | Target Enzyme | Mechanism |
|---|---|---|
| Acyclovir | Viral thymidine kinase → viral DNA polymerase | Selectively activated by HSV/VZV thymidine kinase → acyclovir triphosphate → chain terminator (no 3′-OH) |
| Ganciclovir | CMV kinase → viral DNA polymerase | Similar to acyclovir; broader (CMV); more myelosuppressive |
| Zidovudine (AZT) | HIV reverse transcriptase | dideoxythymidine analogue → incorporated → chain termination; requires cellular kinases |
| Tenofovir | HIV/HBV reverse transcriptase | Acyclic nucleotide phosphonate; long intracellular half-life |
| Ribavirin | Multiple (viral RNA-dependent RNA pol) | Purine nucleoside analogue; inhibits viral capping, RdRp; mutagenic for RNA viruses |
| Remdesivir | Viral RNA-dependent RNA pol | Adenosine analogue → chain termination; used in SARS-CoV-2 |
Anti-Gout Drugs
| Drug | Target | Mechanism |
|---|---|---|
| Allopurinol | Xanthine oxidase | Hypoxanthine analogue → competitive then suicide inhibitor |
| Febuxostat | Xanthine oxidase | Non-purine XO inhibitor; no cross-reactivity with allopurinol |
| Probenecid | URAT1 (renal urate transporter) | Blocks urate reabsorption → increased urinary urate excretion |
| Rasburicase | Uric acid → allantoin | Recombinant uricase; used in tumour lysis syndrome |
| Colchicine | Microtubule polymerisation | Binds tubulin → inhibits neutrophil chemotaxis; prevents NLRP3 assembly |
🔄 Section 9 — Connections to Other Pathways
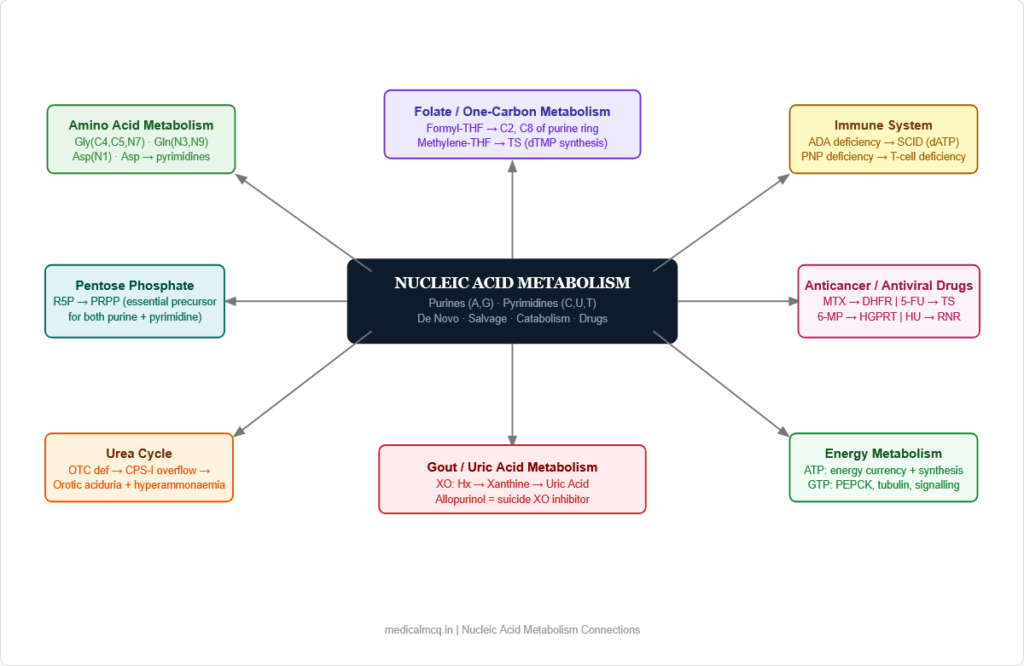
→ Pentose Phosphate Pathway — Ribose-5-phosphate from PPP is the obligatory precursor for PRPP synthesis. Both purine and pyrimidine de novo synthesis depend on PPP as the source of the sugar backbone. G6PD deficiency (PPP blockade) does not directly impair nucleotide synthesis because alternative sources of R5P exist, but the PPP-nucleotide synthesis link is conceptually tested.
→ Amino Acid Metabolism — Glycine, glutamine, and aspartate are essential building blocks of the purine ring. Aspartate also donates nitrogen to pyrimidine synthesis (via carbamoylaspartate). This explains why rapidly proliferating cells have huge demand for these amino acids.
→ One-Carbon Metabolism (Folate) — Formyl-THF donates C2 and C8 of the purine ring (steps 4 and 10 of purine synthesis). Methylene-THF is used by thymidylate synthase for dTMP synthesis. This is why folate deficiency or antifolate drugs (MTX, pemetrexed) simultaneously impair both purine and thymidylate synthesis → megaloblastic anaemia AND impaired DNA replication.
→ TCA Cycle — Fumarate is released during IMP → AMP conversion (adenylosuccinate lyase) — connecting purine synthesis to the TCA cycle. Aspartate for pyrimidine synthesis comes from OAA transamination (TCA intermediate).
→ Energy Metabolism — ATP is both a product and a substrate of nucleotide synthesis (used in multiple steps of both purine and pyrimidine synthesis). The cell’s energy state (ATP/ADP ratio) directly influences nucleotide synthetic rates. Adenine nucleotides are the universal energy currency.
→ Epigenetics / DNA Methylation — SAM (from methionine metabolism) donates methyl groups to cytosine bases in DNA (DNA methyltransferase — DNMT) → 5-methylcytosine. This connects methionine/folate/B12 metabolism directly to gene regulation.
🎯 High-Yield Exam Facts
🔴 Rate-limiting enzyme of de novo purine synthesis = GPAT (glutamine PRPP amidotransferase) at Step 2 Inhibited by all purine end products (AMP, GMP and their phosphorylated forms). PRPP is both substrate and activator.
🔴 Rate-limiting enzyme of de novo pyrimidine synthesis = CPS II (carbamoyl phosphate synthetase II) CPS II is cytoplasmic, uses glutamine. CPS I is mitochondrial, uses NH₃ — for the urea cycle. They are completely different enzymes.
🔴 Purines: built on ribose (ring assembled on sugar). Pyrimidines: ring built first, then attached to ribose (PRPP) This structural difference in synthesis strategy is a direct exam question type.
🔴 Lesch-Nyhan syndrome = HGPRT deficiency = X-linked recessive = compulsive self-mutilation + gout + intellectual disability Allopurinol controls uric acid but does NOT help the neurological symptoms. Pathognomonic for HGPRT deficiency.
🔴 ADA deficiency → dATP accumulates in lymphocytes → SCID (T and B cell failure) First disease treated by gene therapy (1990). PEG-ADA or BMT are current treatments.
🔴 Hereditary Orotic Aciduria = UMP synthase deficiency (OPRT + OMPDC) → megaloblastic anaemia NOT responding to B12/folate Treatment: uridine supplementation. Orotic acid accumulates and is excreted in massive amounts.
🔴 Xanthine oxidase converts Hypoxanthine → Xanthine → Uric Acid (both steps) Allopurinol (hypoxanthine analogue) → converted to oxypurinol → suicide inhibitor of XO. Reduces uric acid in gout and Lesch-Nyhan.
🟠 Purine ring atom donors: Glycine (C4,C5,N7), Glutamine (N3,N9), Aspartate (N1), CO₂ (C6), Formyl-THF (C2,C8) Direct exam recall. “Glycine makes the scaffold” is the memory hook.
🟠 5-FU → FdUMP → irreversible inhibitor of thymidylate synthase (TS) → no dTMP → no DNA synthesis The only source of dTMP is the TS reaction. Block TS = block DNA synthesis in dividing cells.
🟠 Methotrexate inhibits DHFR → depletes active folate (THF) → blocks both dTMP and purine synthesis simultaneously Leucovorin (folinic acid — reduced folate) is the antidote/rescue agent — bypasses DHFR block.
🟠 Gout joint fluid: needle-shaped, negatively birefringent crystals under polarised light Yellow when parallel to compensator. Contrast with pseudogout (CPPD) — rhomboid, weakly positively birefringent.
🟠 Von Gierke disease (G6Pase deficiency) causes hyperuricaemia via two mechanisms:
- Excess PRPP (G6P → PPP → R5P → PRPP → de novo purine overproduction)
- Lactic acidosis (lactate competes with urate for renal excretion → underexcretion)
🟡 CPS I (mitochondrial, NH₃, urea cycle) vs CPS II (cytoplasmic, glutamine, pyrimidine synthesis) — NOT the same enzyme One of the most tested distinctions in nucleotide biochemistry. Ornithine transcarbamylase (OTC) deficiency causes orotic aciduria too — but via carbamoyl phosphate overflow into pyrimidine pathway (not UMP synthase deficiency).
🟡 DHODH (step 4 of pyrimidine synthesis) is the only step on the inner mitochondrial membrane Inhibited by leflunomide/teriflunomide — used in RA and MS. All other pyrimidine synthesis steps are cytoplasmic (first 3 by CAD complex).
🟡 Urate-lowering drugs: Allopurinol/Febuxostat (↓production) vs Probenecid (↑excretion) Probenecid is contraindicated in patients with renal stones (increases urinary urate → more stones). Rasburicase converts urate to allantoin — used in tumour lysis syndrome but contraindicated in G6PD deficiency.
🟡 Ribonucleotide reductase (RNR) converts NDPs → dNDPs — the only source of deoxyribonucleotides Inhibited by hydroxyurea, gemcitabine. This enzyme is why rapidly dividing cells are more sensitive to these drugs.
🧠 Mnemonics & Memory Tricks
“CUT the PY — PURe As Gold” → Helps remember the base family classification
CUT the PY = Cytosine, Uracil, Thymine are PYrimidines PURe As Gold = Adenine and Guanine are PURines
💡 Pro tip: Any question asking “which base is a purine?” — if it’s not C, U, or T, it’s a purine.
“GGAC BFF” — Atom donors for the purine ring (in ring position order) → Simplified version:
Glycine = C4, C5, N7 (the backbone) Glutamine = N3, N9 (two nitrogens) Aspartate = N1 CO₂ = C6 Formyl-THF = C2 (step 4) Formyl-THF = C8 (step 10)
💡 Pro tip: “Two formyl-THF steps = two steps where folate is essential in purine synthesis.” Methotrexate blocks both of these by depleting THF.
“OTC causes Orotic aciduria + UMP synthase causes Orotic aciduria — BUT different mechanisms” → OTC (Ornithine Transcarbamylase) deficiency: Carbamoyl phosphate accumulates → spills into pyrimidine pathway → UMP synthase overwhelmed → orotic acid in urine + hyperammonaemia (urea cycle blocked) → UMP synthase deficiency: Orotic acid cannot be converted → accumulates + megaloblastic anaemia
💡 Pro tip: OTC deficiency = orotic aciduria + HYPERAMMONAEMIA (urea cycle issue). UMP synthase deficiency = orotic aciduria + MEGALOBLASTIC ANAEMIA (no pyrimidines) + normal ammonia.
“AlloXanthine is a Suicide — Allopurinol kills XO” → Allopurinol (hypoxanthine analogue) → converted by XO to oxypurinol (alloxanthine) → oxypurinol binds permanently to XO → suicide inhibition → Drug becomes the weapon; XO destroys itself in the process
💡 Pro tip: “Suicide inhibitor” means the enzyme activates the drug into an active form, which then irreversibly inactivates the enzyme. 5-FU → FdUMP → suicide inhibitor of thymidylate synthase. Allopurinol → oxypurinol → suicide inhibitor of XO.
⚠️ Common Mistakes Students Make on This Topic
❌ Mistake: “CPS I and CPS II are the same enzyme in different compartments” ✅ Reality: CPS I (mitochondrial) uses free NH₃ as nitrogen donor, requires N-acetylglutamate (NAG) as activator, and functions in the urea cycle. CPS II (cytoplasmic) uses glutamine as nitrogen donor, is activated by ATP/PRPP, and functions in pyrimidine synthesis. They have completely different substrates, activators, and genetic origins. 📝 How this gets tested: “A patient with OTC deficiency has orotic aciduria — why?” Because OTC deficiency → carbamoyl phosphate accumulates in mitochondria → exits to cytoplasm → overwhelms pyrimidine synthesis → orotic acid builds up. This requires understanding that CPS I (mitochondria, urea cycle) is upstream of the accumulation.
❌ Mistake: “Allopurinol only inhibits the last step of uric acid synthesis” ✅ Reality: Allopurinol inhibits xanthine oxidase, which catalyses TWO steps: Hypoxanthine → Xanthine AND Xanthine → Uric acid. Both steps are blocked. Furthermore, the accumulated xanthine and hypoxanthine are more soluble than uric acid, so they are excreted in urine without forming stones. 📝 How this gets tested: “After allopurinol treatment, which substances increase in urine?” — Xanthine and hypoxanthine (not uric acid).
❌ Mistake: “In Lesch-Nyhan syndrome, allopurinol cures the disease” ✅ Reality: Allopurinol treats the hyperuricaemia (preventing gout and kidney stones) but has absolutely no effect on the neurological and behavioural manifestations (self-mutilation, intellectual disability, choreoathetosis). These are caused by the biochemical consequences of HGPRT deficiency in the brain — not by uric acid itself. 📝 How this gets tested: “A child with Lesch-Nyhan is started on allopurinol — which symptoms will improve?” Only gout and kidney stones — NOT the self-mutilation or neurological features.
❌ Mistake: “Methotrexate only affects DNA synthesis by blocking folate” ✅ Reality: Methotrexate blocks DHFR → depletes THF → impairs BOTH (1) thymidylate synthesis (dTMP — pyrimidine pathway) AND (2) purine synthesis (steps 4 and 10 need formyl-THF). MTX therefore simultaneously depletes pyrimidines AND purines for DNA synthesis. Leucovorin (folinic acid) rescue bypasses DHFR and restores both pathways. 📝 How this gets tested: “Why does leucovorin rescue work for methotrexate toxicity?” — Leucovorin is already reduced (5-formyl-THF) and does not need DHFR to be activated — it feeds directly into the THF pool.
❌ Mistake: “Orotic aciduria always means UMP synthase deficiency” ✅ Reality: Orotic aciduria can be caused by multiple conditions: (1) UMP synthase deficiency (hereditary orotic aciduria — no hyperammonaemia); (2) OTC deficiency (excess carbamoyl phosphate → pyrimidine pathway → orotic acid + severe hyperammonaemia); (3) Allopurinol use (via altered purine metabolism); (4) High dietary protein. The critical distinguishing feature is ammonia level — elevated in OTC deficiency, normal in UMP synthase deficiency. 📝 How this gets tested: Orotic aciduria + hyperammonaemia = OTC deficiency (urea cycle). Orotic aciduria + megaloblastic anaemia + normal ammonia = UMP synthase deficiency.
📝 5 Practice MCQs — Test Yourself Now
Q1: Which enzyme catalyses the committed, rate-limiting step of de novo purine synthesis?
- A. PRPP synthetase
- B. Adenylosuccinate synthetase
- C. Glutamine phosphoribosylpyrophosphate amidotransferase (GPAT)
- D. IMP dehydrogenase
✅ Answer: C. Glutamine PRPP amidotransferase (GPAT)
Why correct: GPAT catalyses Step 2 of de novo purine synthesis: PRPP + Glutamine → 5-phosphoribosylamine (PRA). This is the committed, irreversible step that determines the rate of the entire pathway. It is allosterically inhibited by all purine nucleotides (AMP, GMP and their phosphates) — classic feedback inhibition.
Why A is wrong: PRPP synthetase catalyses PRPP formation (Step 1 — prerequisite) and is regulated, but it is not the committed step because PRPP is used by BOTH purine and pyrimidine synthesis (and histidine synthesis). The committed step for purines specifically is Step 2. Why B is wrong: Adenylosuccinate synthetase catalyses IMP → Adenylosuccinate (the branch toward AMP) — a late step, not the rate-limiting committed step. Why D is wrong: IMP dehydrogenase catalyses IMP → XMP (the branch toward GMP) — another late step. Inhibited by GMP for feedback, but not the rate-limiting enzyme of the whole pathway.
Exam tip: Three major rate-limiting committed enzymes to know: GPAT (purines), CPS II (pyrimidines), PRPP synthetase (prerequisite but not committed). Any question saying “rate-limiting of de novo purine synthesis” = GPAT.
Q2: A 5-year-old boy presents with recurrent joint swelling, orange-tinged crystals in his nappies from infancy, intellectual disability, and a peculiar behaviour of biting his fingers until they bleed. Uric acid is 12 mg/dL. Allopurinol is started. Which symptoms will NOT improve with this treatment?
- A. Joint swelling and gout attacks
- B. Uric acid crystal deposition in kidneys
- C. Self-mutilating behaviour and choreoathetosis
- D. Serum uric acid levels
✅ Answer: C. Self-mutilating behaviour and choreoathetosis
Why correct: This is Lesch-Nyhan syndrome (HGPRT deficiency, X-linked recessive). Allopurinol inhibits xanthine oxidase → reduces uric acid production → effectively controls hyperuricaemia and its consequences (gout, nephrolithiasis, nephropathy). However, the neurological and behavioural manifestations of Lesch-Nyhan — including the compulsive self-mutilation, choreoathetosis, dystonia, and intellectual disability — are caused by HGPRT deficiency’s biochemical effects in the brain (likely related to abnormal dopaminergic pathways) and are NOT caused by uric acid accumulation itself. Allopurinol has no effect on these features.
Why A is wrong: Joint swelling and gout attacks are caused by MSU crystal deposition, which is directly driven by hyperuricaemia. Allopurinol reduces uric acid → reduces crystal formation → improves gout attacks. Why B is wrong: Uric acid nephropathy and nephrolithiasis respond to allopurinol because crystal deposition requires supersaturated urate — reducing urate production prevents new crystal formation. Why D is wrong: Serum uric acid falls dramatically with allopurinol — this is the intended pharmacological effect.
Exam tip: Lesch-Nyhan syndrome = HGPRT deficiency + self-mutilation + gout + intellectual disability. Allopurinol fixes the gout. Nothing fixes the brain. The self-mutilation is pathognomonic — no other disorder causes this specific behavioural pattern.
Q3: A 3-year-old girl has recurrent bacterial and viral infections, failure to thrive, and lymphopenia. Immune workup shows absent T cells and markedly reduced B cells. The paediatrician suspects an inborn error of metabolism. The accumulation of which metabolite is responsible for the lymphocyte destruction?
- A. Orotic acid
- B. Uric acid
- C. dATP
- D. Xanthine
✅ Answer: C. dATP
Why correct: This is ADA (Adenosine Deaminase) deficiency causing SCID. ADA normally converts adenosine → inosine and deoxyadenosine → deoxyinosine. Without ADA, deoxyadenosine accumulates and is phosphorylated to dATP. High dATP: (1) inhibits ribonucleotide reductase → blocks DNA synthesis in lymphocytes; (2) induces apoptosis in developing T and B lymphocytes; (3) is particularly toxic to lymphocytes because they have high kinase and low 5′-nucleotidase activity (they preferentially phosphorylate rather than dephosphorylate nucleosides). The result is selective destruction of lymphocytes → combined T and B cell immunodeficiency (SCID).
Why A is wrong: Orotic acid accumulation occurs in hereditary orotic aciduria (UMP synthase deficiency) and OTC deficiency — these cause megaloblastic anaemia and urea cycle dysfunction, not immunodeficiency. Why B is wrong: Uric acid accumulation causes gout and Lesch-Nyhan syndrome — not lymphocyte destruction or SCID. Why D is wrong: Xanthine accumulation occurs with xanthine oxidase deficiency (xanthinuria) — causes xanthine kidney stones but not immunodeficiency.
Exam tip: ADA deficiency → dATP accumulation → SCID (T + B cell failure). PNP deficiency → dGTP accumulation → primarily T cell deficiency (B cells relatively spared). Both are purine nucleoside metabolism defects, but with distinct immunological phenotypes.
Q4: A woman diagnosed with rheumatoid arthritis is started on leflunomide. Her rheumatologist explains that the active metabolite of leflunomide specifically targets pyrimidine synthesis. Which enzyme and which step of pyrimidine synthesis is inhibited?
- A. CPS II — Step 1
- B. Aspartate transcarbamoylase — Step 2
- C. Dihydroorotate dehydrogenase (DHODH) — Step 4
- D. UMP synthase — Steps 5 and 6
✅ Answer: C. Dihydroorotate dehydrogenase (DHODH) — Step 4
Why correct: Leflunomide is converted to its active metabolite A771726 (teriflunomide), which inhibits DHODH — the enzyme that catalyses dihydroorotate → orotate (Step 4 of pyrimidine synthesis). DHODH is uniquely located on the inner mitochondrial membrane (the only pyrimidine synthesis enzyme not in the cytoplasm). By blocking DHODH, leflunomide prevents rapidly proliferating lymphocytes (which depend on de novo pyrimidine synthesis for DNA replication) from producing sufficient pyrimidines — providing immunosuppression in RA. Teriflunomide is also approved for multiple sclerosis.
Why A is wrong: CPS II (Step 1) is the rate-limiting enzyme, inhibited by UTP feedback — it is not the target of leflunomide. Why B is wrong: ATCase (Step 2) is the classic allosteric enzyme studied in E. coli — inhibited by CTP, activated by ATP — but it is not the target of leflunomide in clinical medicine. Why D is wrong: UMP synthase (Steps 5–6) deficiency causes hereditary orotic aciduria, but it is not the target of leflunomide.
Exam tip: Leflunomide/teriflunomide = DHODH inhibitor = Step 4 = mitochondrial membrane = pyrimidine synthesis. This is tested in pharmacology sections alongside the disease (RA, MS) and in biochemistry sections asking about which step of pyrimidine synthesis is on the mitochondrial membrane.
Q5: A child presents with megaloblastic anaemia that does not respond to either vitamin B12 or folic acid supplementation. Urinary orotic acid is markedly elevated. Which treatment is most appropriate?
- A. Adenosine supplementation
- B. Allopurinol
- C. Uridine supplementation
- D. High-dose cyanocobalamin (B12)
✅ Answer: C. Uridine supplementation
Why correct: This is hereditary orotic aciduria from UMP synthase deficiency (OPRT + OMPDC). Orotic acid accumulates because it cannot be converted to OMP and then UMP. The result is a deficiency of all pyrimidines (UMP, CMP, dTMP) → cells cannot synthesise DNA → megaloblastic anaemia. The anaemia does NOT respond to B12 or folate (it is not a one-carbon metabolism problem — the folate pathway is intact). Treatment: uridine supplementation — uridine is phosphorylated by uridine kinase to UMP, bypassing the defective UMP synthase. Uridine also feeds back to inhibit CPS II → reduces orotic acid overproduction.
Why A is wrong: Adenosine supplementation would provide purines, but the defect is in pyrimidine synthesis. Adenosine would not address the UMP/CMP/TMP deficiency causing the megaloblastic anaemia. Why B is wrong: Allopurinol inhibits xanthine oxidase → reduces uric acid. It has no role in pyrimidine synthesis or orotic aciduria from UMP synthase deficiency. (Allopurinol can cause orotic aciduria as a side effect, but that is a different mechanism.) Why D is wrong: High-dose B12 is appropriate for B12-deficient megaloblastic anaemia with subacute combined degeneration. In this case, the anaemia is explicitly stated to not respond to B12, and the cause is a pyrimidine synthesis defect, not a cobalamin deficiency.
Exam tip: Orotic aciduria + megaloblastic anaemia + does NOT respond to B12 or folate = UMP synthase deficiency = hereditary orotic aciduria = treat with URIDINE. If orotic aciduria + hyperammonaemia = OTC deficiency (urea cycle) = treat with protein restriction + arginine/citrulline + nitrogen scavengers.
📚 References
📖 Harper’s Illustrated Biochemistry — Rodwell et al. | Chapter 33: Metabolism of Purine & Pyrimidine Nucleotides
📖 Lippincott’s Illustrated Reviews: Biochemistry — Ferrier | Chapter 22: Nucleotide Metabolism
📖 Lehninger Principles of Biochemistry — Nelson & Cox | Chapter 28: Nucleotide Biosynthesis
📖 Stryer’s Biochemistry — Berg, Tymoczko & Stryer | Chapter 25: Nucleotide Biosynthesis
📖 Vasudevan & Sreekumari’s Textbook of Biochemistry — Vasudevan | Chapter 14: Nucleotide Metabolism
📖 Katzung’s Basic & Clinical Pharmacology — Katzung | Chapter 54: Cancer Chemotherapy; Chapter 49: Antiviral Agents
🚀 Keep Practising — You Are Not Done Yet
Nucleic acid metabolism questions in NEET PG and USMLE combine biochemistry with pharmacology and clinical medicine unlike almost any other topic. The question about a child with self-mutilation is testing HGPRT. The question about 5-FU toxicity is testing thymidylate synthase. The question about a child with recurrent infections after a fever is testing ADA deficiency. Knowing the pathway is the bridge between the clinical picture and the correct answer.
medicalmcq.in has free Biochemistry MCQs on purine synthesis, pyrimidine synthesis, salvage pathways, gout pharmacology, and anti-cancer drug mechanisms — all in clinical-scenario format with detailed explanations.
Do the nucleotide metabolism MCQ set immediately. The pattern recognition for these diseases is something that builds with practice, not re-reading.