What You Will Learn in This Article
- Trace the complete pathway of fatty acid synthesis from Acetyl-CoA to palmitate, naming every enzyme and cofactor
- Trace beta-oxidation step by step, calculating the exact ATP yield for any saturated fatty acid
- Explain ketogenesis — where it happens, when it happens, and the 4-step pathway to ketone bodies
- State the rate-limiting enzymes for synthesis, oxidation, and ketogenesis and know what regulates each
- Identify the key differences between fatty acid synthesis and beta-oxidation (location, carriers, cofactors)
- Connect lipid metabolism to glycolysis, the TCA cycle, and amino acid metabolism
- Explain the clinical basis of diabetic ketoacidosis, fatty liver, essential fatty acid deficiency, and lipoprotein disorders
- Answer clinical-scenario MCQs on lipid metabolism with confidence
📖 Introduction: Why This Topic Matters in Exams
A 45-year-old poorly controlled diabetic is brought to the emergency department with deep, laboured breathing (Kussmaul respiration), fruity breath odour, and a blood glucose of 480 mg/dL. Blood gases show a wide anion gap metabolic acidosis. Urine is strongly positive for ketones. This is diabetic ketoacidosis — and every single feature of this presentation is a direct consequence of runaway lipid metabolism. Without insulin, glucose cannot enter cells, so the body mobilises fatty acids from adipose tissue at an uncontrolled rate. The liver, flooded with fatty acids, converts them to ketone bodies faster than peripheral tissues can consume them. Acids accumulate. The patient deteriorates. Understanding lipid metabolism is not abstract biochemistry — it is the molecular basis of one of the most common metabolic emergencies you will manage as a doctor.
Lipid metabolism is among the highest-yield topics in NEET PG and USMLE Step 1 biochemistry. Questions test it at every level: pure recall (“which enzyme is rate-limiting in fatty acid synthesis?”), mechanistic reasoning (“why does malonyl-CoA inhibit carnitine acyltransferase-I?”), and clinical application (“a patient on a prolonged fast develops hypoketotic hypoglycaemia — which enzyme defect is responsible?”). It also connects to pharmacology (statins, fibrates, niacin), pathology (atherosclerosis, fatty liver, DKA), and physiology (fed vs. fasted state regulation).
This article covers the full scope: fatty acid synthesis, beta-oxidation, ketogenesis, lipoprotein metabolism, cholesterol synthesis, and their regulation — along with clinical disorders, high-yield exam facts, mnemonics, and five practice MCQs. Work through it section by section.
🔬 Section 1 — Fatty Acid Synthesis: Building Fat from Acetyl-CoA
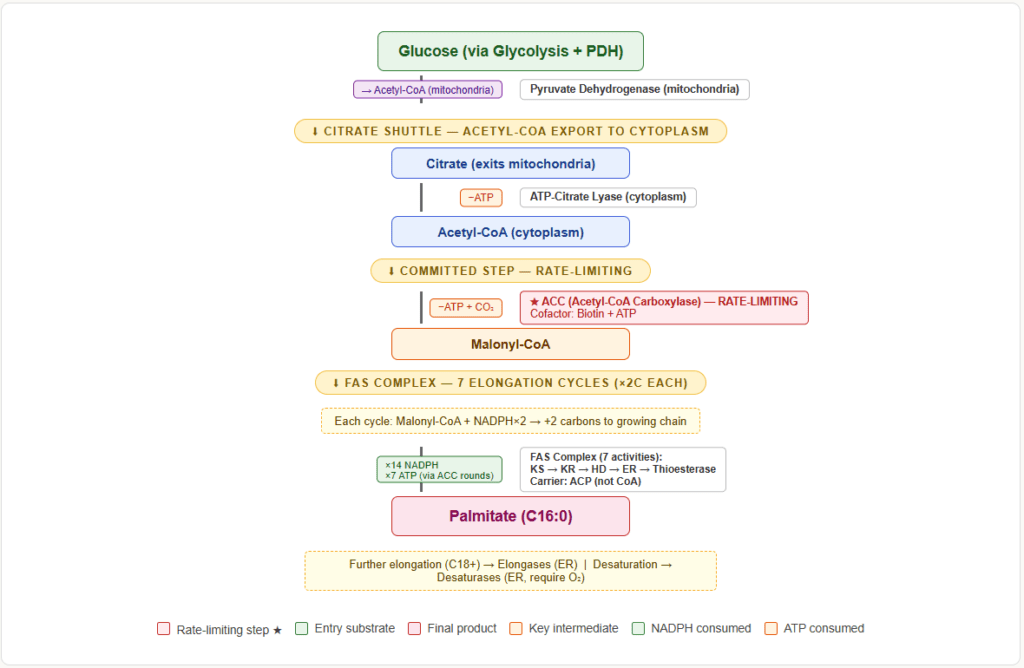
Where It Happens and When
Fatty acid synthesis (de novo lipogenesis) occurs in the cytoplasm (cytosol). It is active in the fed state when insulin is high and glucose is abundant. The primary sites are:
- Liver (most active)
- Adipose tissue
- Lactating mammary glands
- Brain (to a limited extent)
The raw material is Acetyl-CoA, but Acetyl-CoA is produced inside the mitochondria (from pyruvate dehydrogenase and beta-oxidation). It cannot cross the inner mitochondrial membrane directly. So the cell uses a transport trick: Acetyl-CoA condenses with oxaloacetate (OAA) to form citrate, which exits the mitochondria via the citrate shuttle. In the cytosol, ATP-citrate lyase cleaves citrate back into Acetyl-CoA and OAA.
Step-by-Step: From Acetyl-CoA to Palmitate
Fatty acid synthesis builds a 16-carbon fatty acid (palmitate) by adding 2-carbon units sequentially. Each round of elongation requires one Acetyl-CoA, one malonyl-CoA, and 2 NADPH.
Committed Step — Acetyl-CoA → Malonyl-CoA
- Enzyme: Acetyl-CoA carboxylase (ACC) — the rate-limiting enzyme of fatty acid synthesis
- Cofactor: Biotin (CO₂ carrier), ATP, Mn²⁺
- Reaction: Acetyl-CoA + CO₂ + ATP → Malonyl-CoA + ADP + Pi
- This is the irreversible, committed step. Everything that follows depends on malonyl-CoA availability.
The Fatty Acid Synthase (FAS) Complex — 7 Enzymatic Activities in One Protein
In mammals, all 7 steps of the elongation cycle are carried out by a single multifunctional enzyme: Fatty Acid Synthase (FAS). The growing fatty acid chain is attached to an Acyl Carrier Protein (ACP) domain, which carries it between active sites.
The FAS cycle (one round = adds 2 carbons):
| Step | Reaction | Enzyme Activity on FAS |
|---|---|---|
| 1 — Loading | Acetyl-CoA + ACP → Acetyl-ACP | Acetyl transacylase |
| 2 — Loading | Malonyl-CoA + ACP → Malonyl-ACP | Malonyl transacylase |
| 3 — Condensation | Acetyl-ACP + Malonyl-ACP → Acetoacetyl-ACP + CO₂ | β-ketoacyl-ACP synthase (KS) |
| 4 — Reduction | Acetoacetyl-ACP + NADPH → β-hydroxybutyryl-ACP | β-ketoacyl-ACP reductase |
| 5 — Dehydration | β-hydroxybutyryl-ACP → Enoyl-ACP + H₂O | β-hydroxyacyl-ACP dehydratase |
| 6 — Reduction | Enoyl-ACP + NADPH → Butyryl-ACP | Enoyl-ACP reductase |
| 7 — Release (final) | Palmitoyl-ACP → Palmitate + ACP | Thioesterase (after 7 cycles) |
After 7 complete cycles, the chain is 16 carbons long — palmitate (16:0). Thioesterase releases it from ACP as free palmitate.
Overall equation for palmitate synthesis:
8 Acetyl-CoA + 7 ATP + 14 NADPH + 14 H⁺ → Palmitate + 8 CoA + 7 ADP + 7 Pi + 6 H₂O
Source of NADPH (essential — 14 molecules needed per palmitate):
- Pentose phosphate pathway — major source (glucose-6-phosphate dehydrogenase)
- Malic enzyme (malate → pyruvate + CO₂ + NADPH) — the cytosolic malate dehydrogenase step
- NADPH is NOT interchangeable with NADH — synthesis uses NADPH; beta-oxidation produces NADH and FADH₂
Key Difference: Synthesis vs. Beta-Oxidation Side-by-Side
| Feature | Fatty Acid Synthesis | Beta-Oxidation |
|---|---|---|
| Location | Cytoplasm | Mitochondrial matrix |
| Carrier | Acyl Carrier Protein (ACP) | Coenzyme A (CoA) |
| 2-carbon unit | Malonyl-CoA (activated) | Acetyl-CoA (product) |
| Cofactor for reduction/oxidation | NADPH (consumed) | NADH + FADH₂ (produced) |
| Transport into organelle | Citrate shuttle (Acetyl-CoA out) | Carnitine shuttle (Acyl-CoA in) |
| Rate-limiting enzyme | Acetyl-CoA carboxylase (ACC) | Carnitine acyltransferase-I (CAT-I) |
| Active in fed or fasted? | Fed (insulin high) | Fasted (glucagon/epinephrine high) |
| Inhibitor linking both | Malonyl-CoA inhibits CAT-I — the key link |
⚙️ Section 2 — Beta-Oxidation: Breaking Down Fatty Acids for Energy
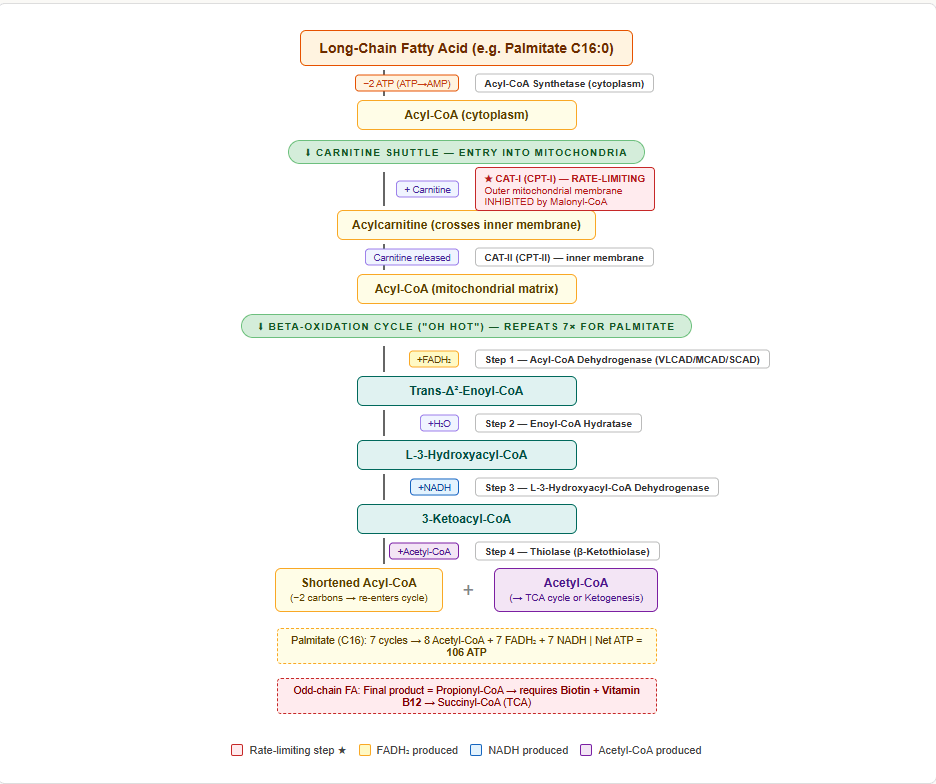
The Carnitine Shuttle — Getting Fatty Acids into the Mitochondria
Long-chain fatty acids cannot cross the inner mitochondrial membrane on their own. They must be transported as acylcarnitine:
- Fatty acid + CoA → Acyl-CoA (in cytoplasm) — enzyme: Acyl-CoA synthetase (uses 2 ATP equivalents: ATP → AMP + 2Pi)
- Acyl-CoA + Carnitine → Acylcarnitine — enzyme: Carnitine acyltransferase-I (CAT-I / CPT-I) — on the outer mitochondrial membrane — rate-limiting step of beta-oxidation
- Acylcarnitine crosses the inner membrane via carnitine-acylcarnitine translocase
- Inside mitochondria: Acylcarnitine + CoA → Acyl-CoA + Carnitine — enzyme: Carnitine acyltransferase-II (CAT-II / CPT-II)
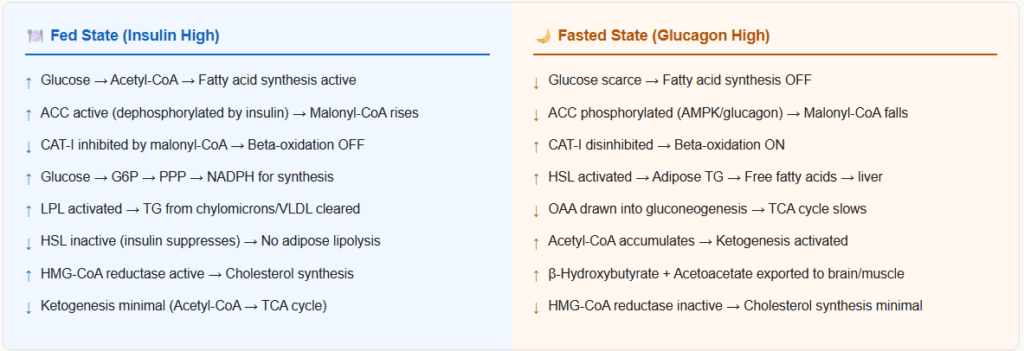
CAT-I is inhibited by malonyl-CoA — this is the critical regulatory link between synthesis and oxidation. When the fed state promotes fatty acid synthesis (malonyl-CoA rises), malonyl-CoA simultaneously blocks fatty acid entry into the mitochondria for oxidation. The cell cannot synthesise and oxidise fat simultaneously — a beautifully efficient switch.
The 4 Steps of One Beta-Oxidation Cycle
Beta-oxidation removes 2 carbons from the fatty acyl chain in each cycle, producing one Acetyl-CoA, one FADH₂, and one NADH.
Step 1 — Oxidation
- Acyl-CoA → Trans-Δ²-enoyl-CoA
- Enzyme: Acyl-CoA dehydrogenase (uses FAD → FADH₂ produced)
- There are three isoforms depending on chain length: VLCAD (very long chain), MCAD (medium chain), SCAD (short chain)
- MCAD deficiency is the most common fatty acid oxidation disorder
Step 2 — Hydration
- Trans-Δ²-enoyl-CoA → L-3-hydroxyacyl-CoA
- Enzyme: Enoyl-CoA hydratase (adds H₂O)
Step 3 — Oxidation
- L-3-hydroxyacyl-CoA → 3-ketoacyl-CoA
- Enzyme: L-3-hydroxyacyl-CoA dehydrogenase (uses NAD⁺ → NADH produced)
Step 4 — Thiolysis
- 3-ketoacyl-CoA → Acyl-CoA (shortened by 2C) + Acetyl-CoA
- Enzyme: Thiolase (β-ketothiolase)
The shortened acyl-CoA re-enters the cycle. Acetyl-CoA enters the TCA cycle.
Memory aid — the 4 steps: Oxidation → Hydration → Oxidation → Thiolysis = “Oh HOT”
Calculating ATP Yield from Beta-Oxidation
For palmitate (C16:0) — a 16-carbon saturated fatty acid:
- Number of beta-oxidation cycles = (16/2) − 1 = 7 cycles
- Each cycle produces: 1 FADH₂ + 1 NADH + 1 Acetyl-CoA
- Total from 7 cycles: 7 FADH₂ + 7 NADH + 8 Acetyl-CoA
- Each Acetyl-CoA in TCA cycle → 10 ATP (modern accounting)
- Each FADH₂ → 1.5 ATP; each NADH → 2.5 ATP
Calculation:
- 8 Acetyl-CoA × 10 = 80 ATP
- 7 FADH₂ × 1.5 = 10.5 ATP
- 7 NADH × 2.5 = 17.5 ATP
- Gross total = 108 ATP
- Subtract activation cost: −2 ATP (ATP → AMP, equivalent to 2 ATP)
- Net ATP from palmitate = 106 ATP
For odd-chain fatty acids: the final product is propionyl-CoA (3C), which is converted via propionyl-CoA carboxylase → methylmalonyl-CoA → succinyl-CoA → TCA cycle. This step requires Vitamin B12 (as adenosylcobalamin) and biotin.
Oxidation of Unsaturated Fatty Acids
Extra enzymes are needed:
- Enoyl-CoA isomerase — for monounsaturated fatty acids (e.g., oleate 18:1)
- 2,4-dienoyl-CoA reductase (requires NADPH) + enoyl-CoA isomerase — for polyunsaturated fatty acids
- Because one FADH₂ is not generated at a double bond position, unsaturated fatty acids yield slightly less ATP than saturated ones of the same chain length
🔥 Section 3 — Ketogenesis: Fuelling the Brain in Starvation
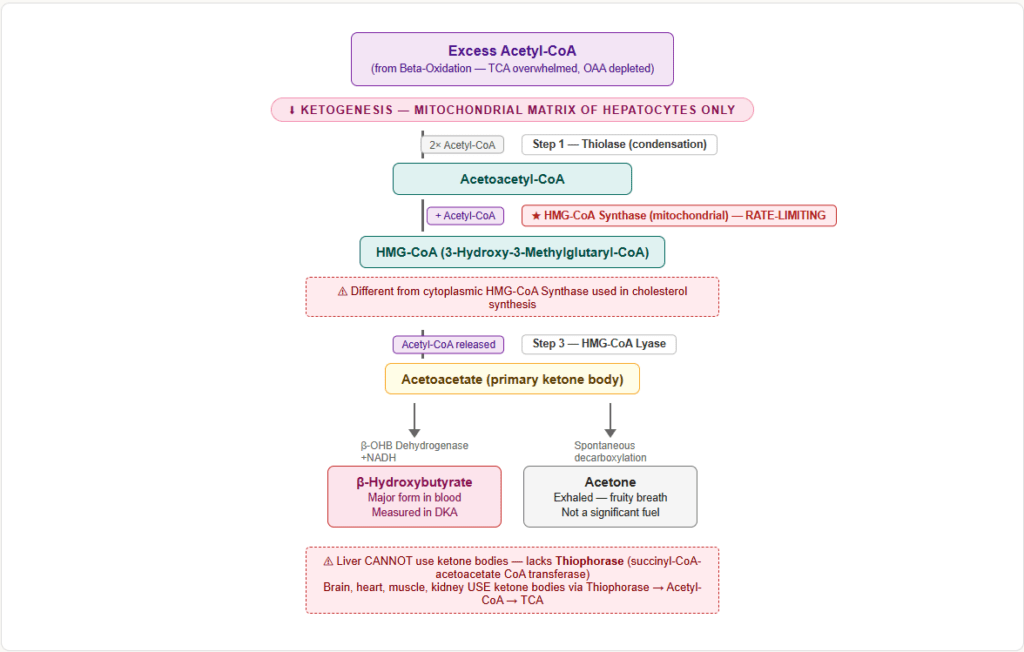
When and Where
Ketogenesis occurs exclusively in the mitochondrial matrix of hepatocytes (liver). It is activated in:
- Prolonged fasting / starvation
- Uncontrolled diabetes mellitus (Type 1)
- High-fat, low-carbohydrate diets
- Prolonged exercise
The trigger: high glucagon, low insulin → adipose tissue releases massive amounts of fatty acids → liver beta-oxidation overwhelms TCA cycle capacity (OAA is diverted to gluconeogenesis) → excess Acetyl-CoA is shunted into ketone body synthesis.
The 4-Step Ketogenesis Pathway
Step 1: 2 Acetyl-CoA → Acetoacetyl-CoA
Enzyme: Thiolase (same enzyme as the last step of beta-oxidation, running in reverse)
Step 2: Acetoacetyl-CoA + Acetyl-CoA → HMG-CoA
Enzyme: HMG-CoA synthase — rate-limiting enzyme of ketogenesis
(This mitochondrial HMG-CoA synthase is different from the cytoplasmic HMG-CoA synthase used in cholesterol synthesis)
Step 3: HMG-CoA → Acetoacetate + Acetyl-CoA
Enzyme: HMG-CoA lyase
Step 4: Acetoacetate → β-hydroxybutyrate (major ketone in blood)
Enzyme: β-hydroxybutyrate dehydrogenase (uses NADH → NAD⁺)
OR: Acetoacetate → Acetone (spontaneous decarboxylation — the fruity breath odour)
The Three Ketone Bodies
| Ketone Body | Notes |
|---|---|
| Acetoacetate | Primary ketone produced; used directly by brain, heart, skeletal muscle |
| β-Hydroxybutyrate | Major form in blood during severe DKA (not technically a ketone — it is a hydroxy acid); measured in labs |
| Acetone | Exhaled; causes fruity breath; not a significant energy source |
Ketone Body Utilisation (Peripheral Tissues, NOT Liver)
The liver produces ketone bodies but cannot use them — it lacks the enzyme succinyl-CoA-acetoacetate CoA transferase (thiophorase). Peripheral tissues (brain, heart, muscle, kidney) use ketones as follows:
- β-Hydroxybutyrate → Acetoacetate (β-hydroxybutyrate dehydrogenase)
- Acetoacetate + Succinyl-CoA → Acetoacetyl-CoA + Succinate — enzyme: Thiophorase (absent in liver)
- Acetoacetyl-CoA → 2 Acetyl-CoA (thiolase)
- Acetyl-CoA → TCA cycle → ATP
🧬 Section 4 — Cholesterol Synthesis (Mevalonate Pathway)
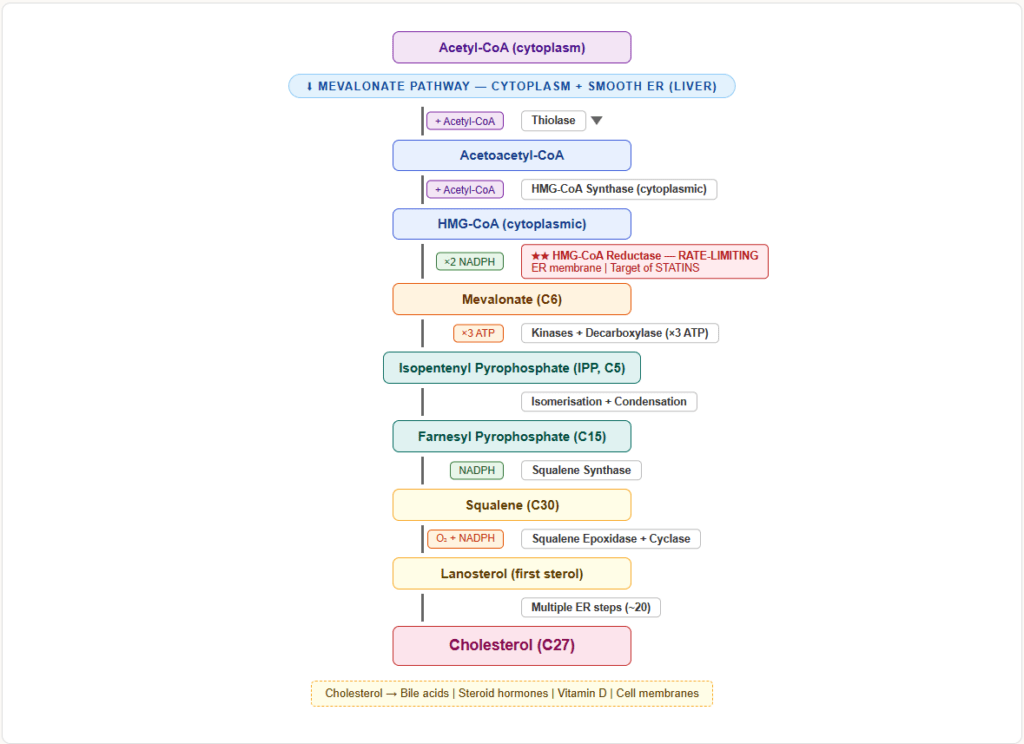
Location and Overview
Cholesterol synthesis occurs in the cytoplasm and smooth ER, primarily in the liver. The pathway starts from Acetyl-CoA and proceeds through the mevalonate pathway.
Key Steps
Step 1: 2 Acetyl-CoA → Acetoacetyl-CoA (thiolase)
Step 2: Acetoacetyl-CoA + Acetyl-CoA → HMG-CoA (HMG-CoA synthase — cytoplasmic isoform)
Step 3: HMG-CoA → Mevalonate — enzyme: HMG-CoA reductase — rate-limiting enzyme of cholesterol synthesis
This step uses 2 NADPH and is the target of statins (competitive inhibitors of HMG-CoA reductase).
Step 4 onwards: Mevalonate → Isopentenyl pyrophosphate → Geranyl-PP → Farnesyl-PP → Squalene → Lanosterol → Cholesterol
Regulation of HMG-CoA Reductase
| Increases activity | Decreases activity |
|---|---|
| Insulin | Glucagon |
| Thyroid hormone (T3) | Glucocorticoids |
| High intracellular cholesterol (SREBP pathway downregulated) — wait, this decreases | Statins (competitive inhibition) |
| Low intracellular cholesterol → SREBP activation → increased transcription | High intracellular cholesterol → SREBP retained in ER → reduced transcription |
Important: HMG-CoA reductase is active in dephosphorylated form (insulin activates phosphatase → dephosphorylates → active). Glucagon/AMP kinase phosphorylates it → inactive.
🚛 Section 5 — Lipoprotein Metabolism
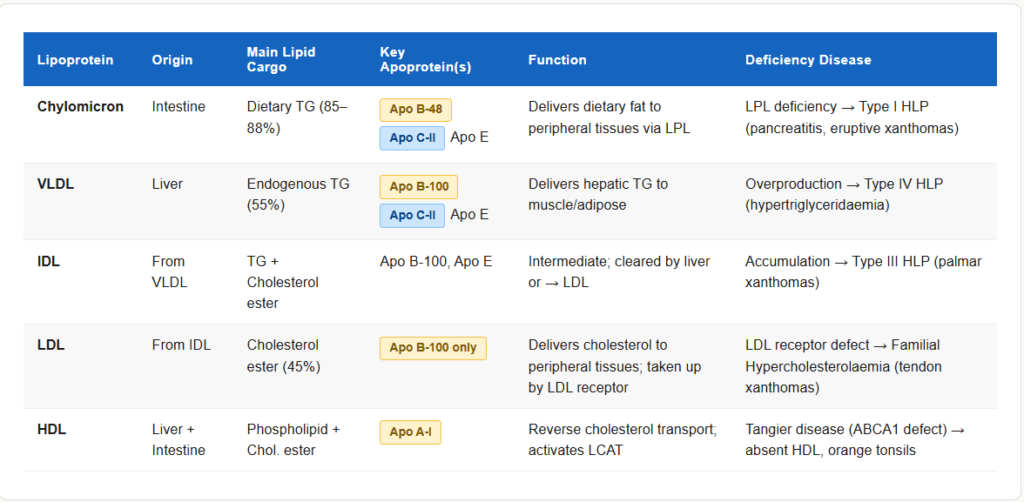
The Lipoprotein Family
Lipids are insoluble in water and must be transported in the blood as lipoproteins — particles with a lipid core and a protein coat (apoproteins).
| Lipoprotein | Origin | Main Cargo | Key Apoprotein | Function |
|---|---|---|---|---|
| Chylomicron | Intestinal epithelium | Dietary TG (exogenous) | Apo B-48, Apo C-II, Apo E | Delivers dietary fat to peripheral tissues |
| VLDL | Liver | Endogenous TG | Apo B-100, Apo C-II, Apo E | Delivers hepatic TG to peripheral tissues |
| IDL | From VLDL (partial lipolysis) | TG + Cholesterol | Apo B-100, Apo E | Intermediate; taken up by liver or becomes LDL |
| LDL | From IDL (further lipolysis) | Cholesterol ester | Apo B-100 only | Delivers cholesterol to peripheral tissues |
| HDL | Liver + Intestine | Phospholipid + Cholesterol | Apo A-I | Reverse cholesterol transport |
Key Enzymes in Lipoprotein Metabolism
Lipoprotein lipase (LPL)
- Located on capillary endothelium of muscle and adipose tissue
- Activated by Apo C-II (from HDL)
- Hydrolyses TG in chylomicrons and VLDL → releases free fatty acids for uptake
- Inhibited by Apo C-III
- Deficiency → Type I hyperlipoproteinaemia (massive hypertriglyceridaemia, eruptive xanthomas, pancreatitis)
Hepatic lipase
- On hepatic sinusoidal endothelium
- Converts IDL → LDL (removes remaining TG)
LCAT (Lecithin-Cholesterol Acyltransferase)
- Activated by Apo A-I
- Esterifies free cholesterol in HDL → cholesterol ester (makes HDL mature)
- Deficiency → fish-eye disease (corneal opacities, low HDL)
CETP (Cholesterol Ester Transfer Protein)
- Transfers cholesterol esters from HDL to VLDL/LDL in exchange for TG
- Target of some experimental drugs (anacetrapib, torcetrapib)
Reverse Cholesterol Transport
HDL scavenges cholesterol from peripheral tissues and returns it to the liver:
- Nascent HDL (disc-shaped) picks up free cholesterol from tissues via ABCA1 transporter
- LCAT (activated by Apo A-I) esterifies cholesterol → HDL becomes spherical (mature)
- CETP transfers cholesterol esters to LDL/VLDL
- HDL delivers cholesterol to liver via SR-BI receptor
Tangier disease — ABCA1 transporter defect → cholesterol cannot leave cells → HDL nearly absent → cholesterol accumulates in tissues (orange tonsils, hepatosplenomegaly, peripheral neuropathy)
🏥 Section 6 — Clinical Connections: When Lipid Metabolism Fails
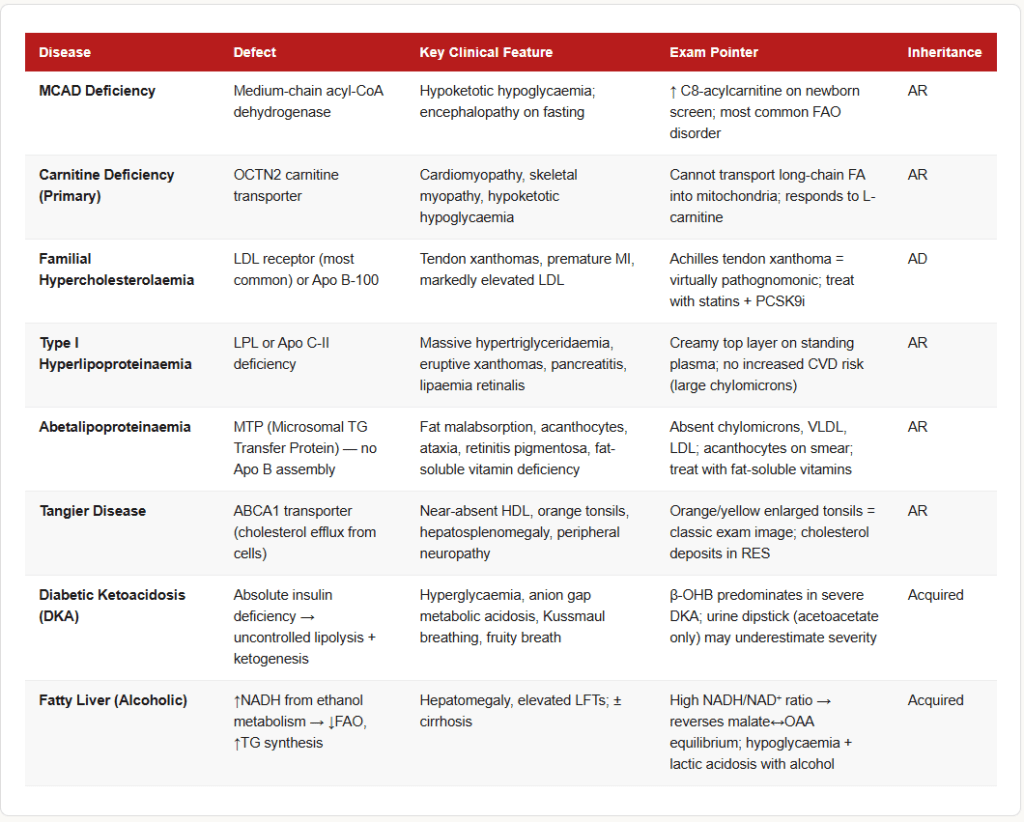
Diabetic Ketoacidosis (DKA)
- Cause: Absolute insulin deficiency (Type 1 DM) or severe insulin deficiency (Type 2 DM)
- Mechanism: No insulin → HSL (hormone-sensitive lipase) is not suppressed → massive lipolysis → hepatic fatty acids flood the liver → beta-oxidation overwhelms TCA capacity (OAA diverted to gluconeogenesis) → Acetyl-CoA accumulates → ketogenesis activated uncontrollably
- Presentation: Hyperglycaemia, ketones in blood/urine, anion gap metabolic acidosis, Kussmaul breathing, fruity breath
- Key metabolic fact: In DKA, β-hydroxybutyrate predominates over acetoacetate. Standard urine dipstick measures acetoacetate (not β-hydroxybutyrate) — so urine ketones may be falsely low in severe DKA.
MCAD Deficiency (Medium-Chain Acyl-CoA Dehydrogenase Deficiency)
- Most common fatty acid oxidation disorder
- Autosomal recessive; commonly presents at 3–24 months during an intercurrent illness (fasting stress)
- Mechanism: Cannot oxidise medium-chain fatty acids (C6–C12) → fatty acids accumulate → no ketone production despite fasting (hypoketotic hypoglycaemia) → no alternative fuel for brain
- Presentation: Hypoketotic hypoglycaemia, encephalopathy, hepatomegaly, sudden death in infancy
- Diagnosis: Elevated C8-acylcarnitine (octanoylcarnitine) on newborn screening (tandem mass spectrometry)
- Treatment: Avoid prolonged fasting; carnitine supplementation controversial
Carnitine Deficiency
- Primary: Defect in OCTN2 transporter (kidneys cannot reabsorb carnitine)
- Secondary: Organic acidaemias, renal tubular disorders, valproate therapy
- Result: Cannot transport long-chain fatty acids into mitochondria → cannot oxidise fat → cardiomyopathy, skeletal myopathy, hypoketotic hypoglycaemia
- Treatment: L-carnitine supplementation
Fatty Liver (Hepatic Steatosis)
Multiple causes converge on one mechanism — TG accumulates in hepatocytes:
| Cause | Mechanism |
|---|---|
| Obesity / insulin resistance | ↑ Fatty acid delivery to liver, ↑ de novo lipogenesis |
| Alcohol | ↑ NADH from alcohol metabolism → ↓ FAO, ↑ TG synthesis |
| Protein malnutrition | ↓ Apo B-100 synthesis → VLDL cannot be assembled → TG trapped in liver |
| Tetracycline / valproate | Inhibit mitochondrial FAO |
| Abetalipoproteinaemia | Apo B-48 / B-100 absent → no chylomicrons or VLDL → fat traps in intestine and liver |
Familial Hypercholesterolaemia (FH)
- Defect: LDL receptor mutation (most common) — LDL cannot be cleared from blood
- Inheritance: Autosomal dominant; heterozygotes have LDL ~2× normal; homozygotes ~4–6× normal
- Clinical: Tendon xanthomas (Achilles, extensor tendons), xanthelasma, corneal arcus, premature atherosclerosis/MI
- Treatment: Statins (HMG-CoA reductase inhibitors) ± PCSK9 inhibitors (evolocumab, alirocumab)
Abetalipoproteinaemia
- Defect: Microsomal triglyceride transfer protein (MTP) — cannot assemble Apo B-containing lipoproteins (chylomicrons, VLDL)
- Result: Fat malabsorption, steatorrhoea, fat-soluble vitamin deficiency (Vitamins A, D, E, K)
- Characteristic: Acanthocytosis (spiky RBCs) due to abnormal membrane lipid composition, ataxia, retinitis pigmentosa
Essential Fatty Acid Deficiency
- Essential fatty acids: Linoleic acid (ω-6, 18:2) and α-Linolenic acid (ω-3, 18:3) — cannot be synthesised by humans
- Deficiency: Dermatitis, hair loss, poor wound healing, increased susceptibility to infection
- Found in: vegetable oils (linoleic), flaxseed/fish oils (linolenic)
- Arachidonic acid (ω-6, 20:4) is derived from linoleic acid — precursor for prostaglandins, thromboxanes, leukotrienes
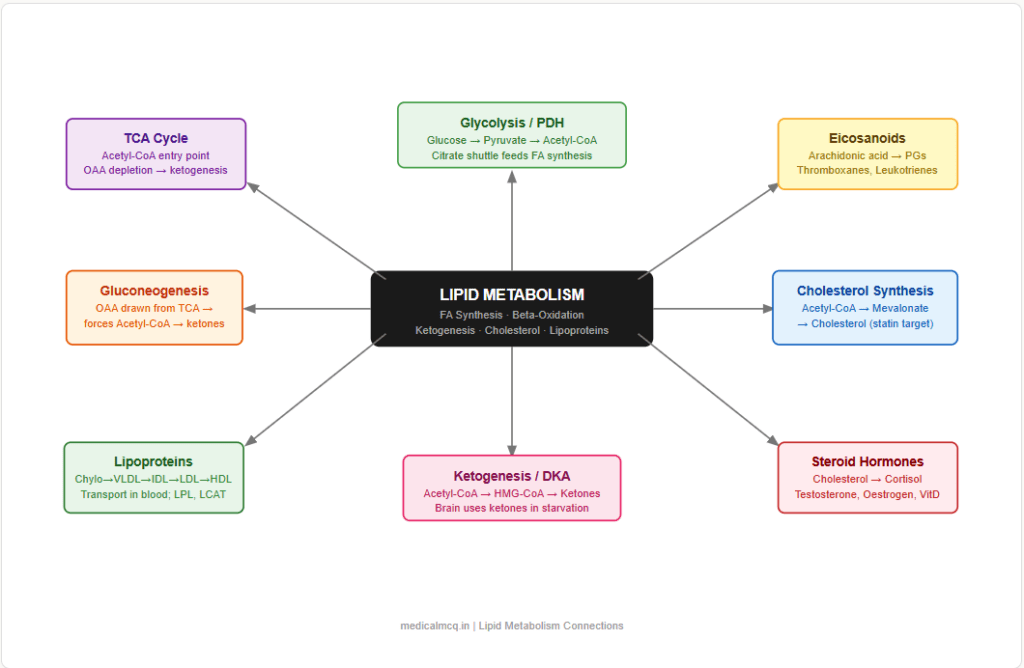
🔄 Section 7 — Connections to Other Metabolic Pathways
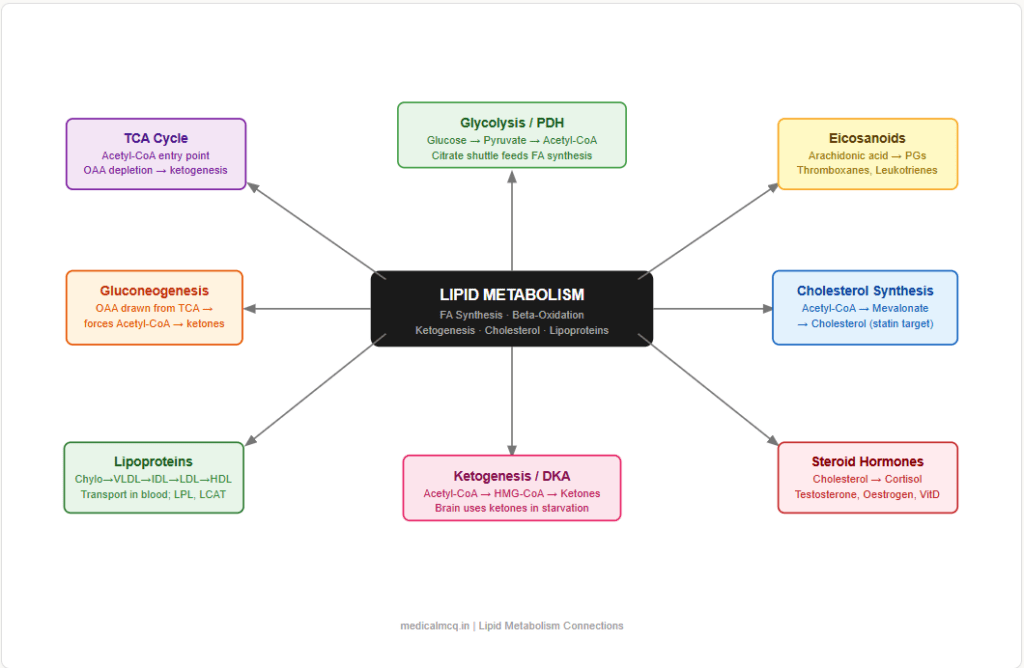
→ Glycolysis — Glucose → Pyruvate → Acetyl-CoA (pyruvate dehydrogenase) → feeds both TCA cycle AND fatty acid synthesis. In the fed state, excess glucose is the carbon source for de novo lipogenesis. Insulin promotes both glucose uptake and fatty acid synthesis simultaneously.
→ TCA Cycle — Acetyl-CoA from beta-oxidation enters the TCA cycle. When TCA is saturated (OAA depleted by gluconeogenesis in fasting), excess Acetyl-CoA goes to ketogenesis instead. Citrate exits mitochondria to provide Acetyl-CoA for cholesterol and fatty acid synthesis in cytoplasm.
→ Gluconeogenesis — In fasting, oxaloacetate is drawn from TCA cycle into gluconeogenesis → TCA cycle slows → Acetyl-CoA cannot be cleared → ketogenesis is forced. Propionyl-CoA from odd-chain FA oxidation provides succinyl-CoA to gluconeogenesis.
→ Amino Acid Metabolism — Ketogenic amino acids (leucine, lysine, phenylalanine, isoleucine, tryptophan, threonine, tyrosine) produce Acetyl-CoA → can form fatty acids or ketone bodies. Glucogenic amino acids → pyruvate or TCA intermediates → OAA → gluconeogenesis.
→ Pentose Phosphate Pathway — The major source of NADPH for fatty acid synthesis. G6PD deficiency impairs NADPH supply, reducing lipogenic capacity.
→ Eicosanoid Synthesis — Arachidonic acid (from membrane phospholipids via phospholipase A2) → prostaglandins (COX pathway), leukotrienes (LOX pathway), thromboxanes. NSAIDs inhibit COX; zileuton inhibits LOX.
→ Steroid Hormones / Bile Acids — Cholesterol is the precursor for all steroid hormones (cortisol, aldosterone, testosterone, oestrogen, progesterone) and bile acids. HMG-CoA reductase is therefore indirectly essential for hormone synthesis.
🎯 High-Yield Exam Facts
These are the exact facts that appear repeatedly across NEET PG, USMLE, AIIMS and FMGE papers.
🔴 ACC (Acetyl-CoA carboxylase) is the rate-limiting enzyme of fatty acid synthesis
Activated by citrate; inhibited by palmitoyl-CoA (product inhibition) and by phosphorylation (glucagon/AMPK). Biotin is the essential cofactor.
🔴 CAT-I (Carnitine acyltransferase-I) is the rate-limiting enzyme of beta-oxidation
Inhibited by malonyl-CoA — this is the key regulatory link that prevents simultaneous synthesis and oxidation of fatty acids.
🔴 HMG-CoA reductase is the rate-limiting enzyme of cholesterol synthesis
Located in the ER; active in dephosphorylated form; inhibited by statins. SREBP regulates its transcription based on intracellular cholesterol levels.
🔴 HMG-CoA synthase (mitochondrial) is the rate-limiting enzyme of ketogenesis
The mitochondrial isoform is for ketogenesis; the cytoplasmic isoform is for cholesterol synthesis. Do not confuse them.
🔴 Fatty acid synthesis occurs in cytoplasm; beta-oxidation occurs in mitochondria
Synthesis uses ACP as carrier; oxidation uses CoA. Synthesis uses NADPH; oxidation produces NADH and FADH₂.
🔴 Malonyl-CoA inhibits CAT-I — the master on/off switch between synthesis and oxidation
In the fed state: insulin → ACC active → malonyl-CoA rises → CAT-I inhibited → no FAO. In fasting: glucagon → ACC phosphorylated (inactive) → malonyl-CoA falls → CAT-I active → FAO proceeds.
🟠 Carnitine is required only for LONG-chain fatty acids (≥C12)
Short and medium-chain fatty acids can enter the mitochondria without carnitine. MCAD deficiency affects medium-chain (C6–C12) oxidation.
🟠 The liver makes ketone bodies but CANNOT use them (lacks thiophorase)
Peripheral tissues (brain, heart, skeletal muscle, kidney) use ketone bodies. After prolonged starvation, the brain can meet up to 75% of its energy needs from ketone bodies.
🟠 Odd-chain fatty acid oxidation yields propionyl-CoA → requires Vitamin B12 and biotin
Propionyl-CoA → (propionyl-CoA carboxylase, biotin) → methylmalonyl-CoA → (methylmalonyl-CoA mutase, B12) → succinyl-CoA. B12 deficiency → methylmalonic aciduria.
🟠 MCAD deficiency presents as hypoketotic hypoglycaemia — not hyperketotic
Cannot oxidise medium-chain fatty acids → no ketone production → brain starves. Elevated C8-acylcarnitine on newborn screen is diagnostic. A common trap: students expect ketosis with fatty acid oxidation defects, but these cause HYPOketosis.
🟠 LDL receptor defect → Familial Hypercholesterolaemia; tendon xanthomas are pathognomonic
Achilles tendon xanthomas = virtually diagnostic of FH. Distinguish from eruptive xanthomas (hypertriglyceridaemia, LPL deficiency).
🟡 β-hydroxybutyrate is the predominant ketone in severe DKA — not acetoacetate
Urine dipstick measures acetoacetate. In very severe DKA, the ratio of β-OHB:acetoacetate shifts heavily towards β-OHB, making urine ketones paradoxically low even as the patient worsens.
🟡 Biotin is required by both ACC (fatty acid synthesis) and propionyl-CoA carboxylase (odd-chain FA oxidation)
Also required by pyruvate carboxylase (gluconeogenesis) and methylcrotonyl-CoA carboxylase. “All carboxylases need biotin.” Biotin deficiency → dermatitis, alopecia, neurological symptoms.
🟡 Apo C-II activates LPL; Apo C-III inhibits LPL
Apo C-II deficiency → hypertriglyceridaemia mimicking LPL deficiency. Apo C-III excess (seen in insulin resistance) → hypertriglyceridaemia.
🟡 Abetalipoproteinaemia → acanthocytes + fat-soluble vitamin deficiency + ataxia
The triad of acanthocytes + retinitis pigmentosa + ataxia in an infant with fat malabsorption = abetalipoproteinaemia (MTP deficiency). Compare with Bassen-Kornzweig syndrome (same disease, older name).
🧠 Mnemonics & Memory Tricks
“Oh HOT!” — The 4 Steps of Beta-Oxidation
→ Helps you remember the sequence and enzyme type for each step
O = Oxidation (Acyl-CoA dehydrogenase → FADH₂)
H = Hydration (Enoyl-CoA hydratase)
O = Oxidation (L-3-hydroxyacyl-CoA dehydrogenase → NADH)
T = Thiolysis (Thiolase → Acetyl-CoA released)
💡 Pro tip: When a question asks “which cofactor is produced first in beta-oxidation?” — the first O is FAD-linked → FADH₂ comes before NADH.
“CHIVL” — Lipoproteins in Order of Decreasing Density (or Increasing Size)
→ Helps you remember: HDL > LDL > IDL > VLDL > Chylomicrons (density order)
Chylomicrons (largest, least dense)
VLDL
IDL
LDL
HDL (smallest, most dense)
💡 Pro tip: Higher density = more protein, less lipid. LDL is “bad” (delivers cholesterol to vessels). HDL is “good” (returns it to liver).
“A Merry King Sells Fine Lavish Cholesterol” — Mevalonate Pathway Steps
→ Helps you remember the sequence from HMG-CoA to Cholesterol
A = Acetyl-CoA (starting material)
M = Mevalonate (from HMG-CoA reductase — rate-limiting, statin target)
K = Mevalonate → Kinase step → Mevalonate-5-phosphate
S = Squalene (farnesyl-PP → squalene via squalene synthase)
F = Lanosterol (squalene → lanosterol — first sterol intermediate)
L = Lanoesterol intermediates
C = Cholesterol
💡 Pro tip: Remember “statins block M = Mevalonate step” — that kills the whole downstream pathway including cholesterol, CoQ10, dolichol, and prenylation.
“Fat CATs Never Oxidise Long chains without Carnitine”
→ Helps you remember that CAT-I (Carnitine Acyl Transferase) is the gatekeeper for LONG-chain FA entry into mitochondria — and that malonyl-CoA inhibits it (the “never” = malonyl-CoA blocks it in the fed state).
💡 Pro tip: “Short and medium chains walk in by themselves — no carnitine needed.” This directly explains why MCAD deficiency and medium-chain triglyceride (MCT) oil therapy work the way they do.
⚠️ Common Mistakes Students Make on This Topic
❌ Mistake: “The rate-limiting enzyme of beta-oxidation is acyl-CoA dehydrogenase”
✅ Reality: The rate-limiting step is CAT-I (carnitine acyltransferase-I) — the transport step. Acyl-CoA dehydrogenase catalyses the first chemical step of the oxidation cycle, but the overall rate is controlled by how much acyl-CoA enters the mitochondria.
📝 How this gets tested: Questions specifically ask for “rate-limiting step of beta-oxidation” — the answer is the carnitine shuttle / CAT-I. Many students confuse the first enzymatic step with the rate-limiting step (the same error they make in glycolysis with hexokinase vs. PFK-1).
❌ Mistake: “Fatty acid synthesis and beta-oxidation are exact reverses of each other”
✅ Reality: They are conceptually opposite but mechanistically very different. Different locations (cytoplasm vs. mitochondria), different carriers (ACP vs. CoA), different cofactors (NADPH vs. FAD/NAD⁺), different activated 2-carbon units (malonyl-CoA vs. Acetyl-CoA). They are regulated in opposite directions by the same signal (malonyl-CoA).
📝 How this gets tested: Tables asking you to identify the correct carrier, cofactor, or location for each process. Choosing CoA for synthesis or ACP for oxidation is the classic wrong answer.
❌ Mistake: “The liver can use ketone bodies for energy”
✅ Reality: The liver cannot use ketone bodies because it lacks thiophorase (succinyl-CoA-acetoacetate CoA transferase). The liver makes ketones exclusively to export them to peripheral tissues.
📝 How this gets tested: “Which tissue cannot utilise ketone bodies?” — the answer is liver (and RBCs, which have no mitochondria). This is the reverse of the usual question pattern.
❌ Mistake: “In DKA, urine dipstick for ketones will always be strongly positive”
✅ Reality: In severe DKA, β-hydroxybutyrate dominates over acetoacetate. Urine dipstick only detects acetoacetate. A patient in severe DKA may have misleadingly low/negative urine ketones on dipstick. β-hydroxybutyrate must be measured directly in blood.
📝 How this gets tested: Clinical vignette of apparent DKA with “negative/trace urine ketones” — the correct interpretation is that β-OHB is predominating, not that ketosis is absent.
❌ Mistake: “All fatty acid oxidation disorders cause ketosis”
✅ Reality: Fatty acid oxidation disorders cause HYPOketotic hypoglycaemia — because the very pathway that produces ketones is defective. This is one of the most important clinical distinctions tested in metabolic disease questions.
📝 How this gets tested: “An infant presents with hypoglycaemia during illness — serum ketones are low/absent” — this is a fatty acid oxidation disorder (MCAD deficiency most commonly), NOT diabetic ketosis.
📝 5 Practice MCQs — Test Yourself Now
Q1: Which enzyme catalyses the rate-limiting, committed step of fatty acid synthesis?
- A. Fatty acid synthase (FAS)
- B. Acetyl-CoA carboxylase (ACC)
- C. ATP-citrate lyase
- D. Malonyl-CoA decarboxylase
✅ Answer: B. Acetyl-CoA carboxylase (ACC)
Why correct: ACC catalyses the carboxylation of Acetyl-CoA to Malonyl-CoA — the committed, irreversible step that determines the rate of the entire fatty acid synthesis pathway. It requires biotin and ATP, and is allosterically activated by citrate and inhibited by palmitoyl-CoA and glucagon-mediated phosphorylation.
Why A is wrong: FAS is a large multifunctional enzyme that carries out the 7-step elongation cycle, but it is downstream of the committed step. FAS activity depends on the supply of malonyl-CoA from ACC.
Why C is wrong: ATP-citrate lyase releases Acetyl-CoA from citrate in the cytoplasm — it is part of the citrate shuttle and enables synthesis, but it is not the rate-limiting step.
Why D is wrong: Malonyl-CoA decarboxylase converts malonyl-CoA back to Acetyl-CoA — it opposes synthesis and is not the rate-limiting enzyme of the synthetic pathway.
Exam tip: “Rate-limiting enzyme of fatty acid synthesis” = ACC. This is asked frequently and directly. Pair it with: “Rate-limiting of beta-oxidation” = CAT-I, and “Rate-limiting of cholesterol synthesis” = HMG-CoA reductase.
Q2: A 14-month-old child is brought to the emergency department after a 24-hour episode of vomiting and reduced oral intake. On examination he is drowsy and hypoglycaemic. Serum ketones are undetectable. Plasma acylcarnitine profile shows elevated C8-acylcarnitine. What is the most likely diagnosis?
- A. Diabetic ketoacidosis
- B. Medium-chain acyl-CoA dehydrogenase (MCAD) deficiency
- C. Pyruvate kinase deficiency
- D. Carnitine deficiency
✅ Answer: B. MCAD deficiency
Why correct: MCAD deficiency impairs oxidation of medium-chain fatty acids (C6–C12). During fasting stress, the child cannot mobilise fat reserves for energy, leading to hypoglycaemia. Because beta-oxidation itself is blocked, Acetyl-CoA cannot be generated, so ketone bodies cannot be formed — hence hypoketotic hypoglycaemia. Elevated C8 (octanoyl) acylcarnitine on newborn screening or acylcarnitine profile is diagnostic.
Why A is wrong: DKA causes HYPERketotic, not hypoketotic, hypoglycaemia, and is associated with insulin deficiency and high blood glucose — not a toddler with vomiting illness.
Why C is wrong: Pyruvate kinase deficiency causes haemolytic anaemia, not a metabolic crisis with hypoketotic hypoglycaemia.
Why D is wrong: Carnitine deficiency could cause hypoketotic hypoglycaemia (long-chain FA cannot enter mitochondria), but the acylcarnitine profile finding of elevated C8 specifically points to MCAD, not carnitine deficiency.
Exam tip: “Hypoketotic hypoglycaemia + intercurrent illness in an infant + elevated C8-acylcarnitine” = MCAD deficiency. This is the most commonly tested fatty acid oxidation disorder.
Q3: In the fed state, malonyl-CoA accumulates in hepatocytes. Which of the following correctly describes the consequence and the mechanism?
- A. Beta-oxidation is stimulated by malonyl-CoA binding to the active site of acyl-CoA dehydrogenase
- B. Beta-oxidation is inhibited because malonyl-CoA inhibits CAT-I, preventing acyl-CoA entry into mitochondria
- C. Fatty acid synthesis is inhibited because malonyl-CoA activates ACC
- D. Ketogenesis is stimulated because malonyl-CoA provides substrate for HMG-CoA synthase
✅ Answer: B. Beta-oxidation is inhibited because malonyl-CoA inhibits CAT-I, preventing acyl-CoA entry into mitochondria
Why correct: Malonyl-CoA allosterically inhibits CAT-I (carnitine acyltransferase-I) on the outer mitochondrial membrane. This prevents long-chain acyl-CoA from being conjugated to carnitine and entering the mitochondria. In the fed state, insulin activates ACC → malonyl-CoA rises → CAT-I is inhibited → fatty acid oxidation is switched off. This is the master switch preventing futile cycling between synthesis and oxidation.
Why A is wrong: Malonyl-CoA does not bind acyl-CoA dehydrogenase. Its target is exclusively CAT-I.
Why C is wrong: Malonyl-CoA is the product of ACC, not an activator. High malonyl-CoA actually provides feedback information but the regulation of ACC is by citrate (activates) and palmitoyl-CoA/phosphorylation (inhibits) — not by malonyl-CoA activating it.
Why D is wrong: Malonyl-CoA is a cytoplasmic molecule with no role in ketogenesis. It is not a substrate for mitochondrial HMG-CoA synthase.
Exam tip: The malonyl-CoA → CAT-I inhibition axis is the single most important regulatory link in lipid metabolism. Expect at least one question on this relationship in any major exam.
Q4: A 35-year-old man presents with tendon xanthomas over the Achilles tendons, corneal arcus, and a serum LDL of 580 mg/dL. His father died of myocardial infarction at age 40. Which is the most likely underlying molecular defect?
- A. LPL deficiency
- B. Apo C-II deficiency
- C. LDL receptor mutation
- D. LCAT deficiency
✅ Answer: C. LDL receptor mutation
Why correct: This is the classic presentation of Familial Hypercholesterolaemia (FH). LDL receptors on hepatocytes normally bind and internalise LDL (via Apo B-100). When these receptors are absent or dysfunctional, LDL cannot be cleared from plasma, leading to massively elevated LDL. Cholesterol deposits in tendons (xanthomas) and the cornea (arcus), and premature atherosclerosis occurs — evidenced here by the father’s early MI.
Why A is wrong: LPL deficiency causes HYPERtriglyceridaemia (eruptive xanthomas, pancreatitis) — not isolated hypercholesterolaemia. LDL is not elevated.
Why B is wrong: Apo C-II deficiency causes hypertriglyceridaemia (similar to LPL deficiency since Apo C-II activates LPL) — not hypercholesterolaemia.
Why D is wrong: LCAT deficiency causes low HDL, fish-eye disease (corneal opacification), and mild hypertriglyceridaemia — not markedly elevated LDL with tendon xanthomas.
Exam tip: Tendon xanthomas (especially Achilles) + markedly elevated LDL + family history of early CAD = Familial Hypercholesterolaemia = LDL receptor defect. This triad appears in virtually every exam.
Q5: A biochemist studying fatty acid metabolism in isolated hepatocytes adds excess palmitate to the culture medium. Which combination of changes in metabolic intermediates would be expected in these cells if they are concurrently deprived of carbohydrate (simulating fasting)?
- A. Malonyl-CoA rises; Acetyl-CoA falls; ketogenesis decreases
- B. Malonyl-CoA falls; CAT-I is disinhibited; Acetyl-CoA accumulates; ketogenesis increases
- C. Malonyl-CoA rises; CAT-I is inhibited; fatty acids accumulate in cytoplasm; ketogenesis decreases
- D. Citrate rises; ACC is activated; malonyl-CoA rises; beta-oxidation decreases
✅ Answer: B. Malonyl-CoA falls; CAT-I is disinhibited; Acetyl-CoA accumulates; ketogenesis increases
Why correct: In carbohydrate deprivation (simulating fasting): insulin falls, glucagon rises → ACC is phosphorylated (inactivated) → malonyl-CoA falls → CAT-I inhibition is relieved → long-chain fatty acids (palmitate) can now enter mitochondria via carnitine shuttle → massive beta-oxidation → Acetyl-CoA accumulates beyond TCA capacity (OAA is also depleted, diverted to gluconeogenesis) → ketogenesis is activated (HMG-CoA synthase, HMG-CoA lyase) → ketone bodies produced. This exactly describes the metabolic state of fasting/DKA.
Why A is wrong: In fasting/carbohydrate deprivation, malonyl-CoA FALLS (ACC is inhibited, not activated). Rising malonyl-CoA is a fed-state response.
Why C is wrong: This describes the fed state (malonyl-CoA rises when carbohydrate is abundant, not when it is removed). Removing carbohydrate does the opposite — malonyl-CoA falls.
Why D is wrong: Citrate rises and ACC is activated in the fed state when glucose is metabolised through glycolysis and TCA, generating abundant citrate. Carbohydrate deprivation starves the citrate shuttle of substrate, so citrate falls.
Exam tip: Integration questions like this test whether you understand the full fed/fasted metabolic switch — not just isolated facts. Draw the regulatory chain: Carbohydrate deprivation → ↓Insulin/↑Glucagon → ↓ACC activity → ↓Malonyl-CoA → ↑CAT-I activity → ↑Beta-oxidation → ↑Acetyl-CoA → ↑Ketogenesis.
📚 References
📖 Harper’s Illustrated Biochemistry — Rodwell et al. | Chapter 22: Oxidation of Fatty Acids; Chapter 23: Biosynthesis of Fatty Acids & Eicosanoids; Chapter 26: Cholesterol Synthesis, Transport & Excretion
📖 Lippincott’s Illustrated Reviews: Biochemistry — Ferrier | Chapter 16: Fatty Acid and Triacylglycerol Metabolism; Chapter 18: Cholesterol and Steroid Metabolism
📖 Lehninger Principles of Biochemistry — Nelson & Cox | Chapter 17: Fatty Acid Catabolism; Chapter 21: Lipid Biosynthesis
📖 Stryer’s Biochemistry — Berg, Tymoczko & Stryer | Chapter 22: Fatty Acid Metabolism; Chapter 26: Biosynthesis of Membrane Lipids and Steroids
📖 Vasudevan & Sreekumari’s Textbook of Biochemistry — Vasudevan | Chapter 11: Metabolism of Lipids
🚀 Keep Practising — You Are Not Done Yet
Lipid metabolism is one of those topics where reading once is never enough. The real exam questions combine two or three concepts from this article — regulation + clinical + ATP yield — in a single scenario.
medicalmcq.in has free Biochemistry MCQs covering every pathway in this article, each with a detailed explanation that builds your reasoning rather than just telling you the answer.
Work through the lipid metabolism set topic by topic — fatty acid synthesis first, then beta-oxidation, then ketogenesis, then lipoproteins. Track where you drop marks. Those gaps are where your exam marks are hiding.