What You Will Learn in This Article
- Name all 8 steps of the Krebs cycle in order with the correct enzyme for each
- State exactly what is produced at each step (NADH, FADH₂, GTP, CO₂) and calculate the total yield per turn
- Identify the 3 irreversible steps and the rate-limiting enzyme
- Explain how the cycle is regulated by energy charge, substrate availability, and key inhibitors
- Connect the TCA cycle to glycolysis, fatty acid oxidation, amino acid metabolism, and gluconeogenesis (anaplerosis)
- Recall the clinical relevance: thiamine deficiency, arsenite poisoning, fluoroacetate poisoning, and enzyme-linked disorders
- Apply rapid exam shortcuts to answer TCA questions in under 30 seconds
📖 Introduction: Why This Topic Matters in Exams
A chronic alcoholic is admitted with confusion, ophthalmoplegia, and ataxia — the classic triad of Wernicke encephalopathy. The cause? Thiamine (Vitamin B1) deficiency. But why does thiamine deficiency wreck the nervous system so profoundly? Because thiamine pyrophosphate (TPP) is the essential cofactor for three key enzymes — pyruvate dehydrogenase, α-ketoglutarate dehydrogenase, and transketolase — and without it, the TCA cycle grinds to a near halt. The brain, which depends almost entirely on oxidative metabolism for its energy, suffers first and fastest. This is not a theoretical question; it appears in clinical-scenario form in virtually every major entrance exam.
The TCA cycle (also called the Krebs cycle or citric acid cycle) is the central hub of cellular metabolism. Everything converges here: carbohydrates via Acetyl-CoA, fatty acids via Acetyl-CoA, and amino acids via multiple entry points. The cycle extracts the chemical energy stored in these molecules as high-energy electron carriers (NADH and FADH₂), which then drive the electron transport chain. Without the TCA cycle, aerobic life is impossible. This makes it one of the highest-yield biochemistry topics in NEET PG and USMLE Step 1.
This article gives you all 8 steps with enzymes, full energy accounting, complete regulation, clinical connections, and — critically — a set of fast exam shortcuts so you can answer TCA questions without hesitation under pressure.
🔬 Section 1 — The 8 Steps of the Krebs Cycle: Every Enzyme, Every Product
Overview: What the Cycle Actually Does
The TCA cycle occurs entirely in the mitochondrial matrix. Each turn of the cycle processes one Acetyl-CoA (2 carbons), which condenses with oxaloacetate (4 carbons) to form citrate (6 carbons). Over 8 steps, 2 carbons are lost as CO₂, the oxaloacetate is regenerated, and the energy is captured as:
- 3 NADH
- 1 FADH₂
- 1 GTP (or ATP)
- 2 CO₂
Since one glucose molecule yields 2 Acetyl-CoA (via 2 pyruvates), the cycle turns twice per glucose.
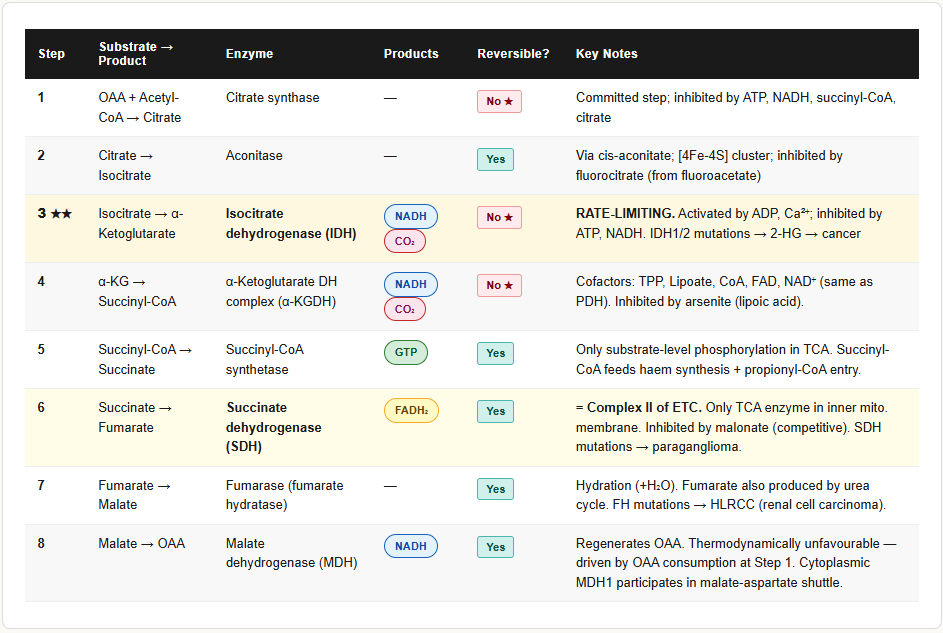
The 8 Steps in Full Detail
STEP 1 — Oxaloacetate + Acetyl-CoA → Citrate
- Enzyme: Citrate synthase
- Reaction type: Condensation
- Irreversible — this is the committed step of the TCA cycle
- Oxaloacetate (4C) + Acetyl-CoA (2C) → Citrate (6C)
- Inhibited by: ATP, NADH, succinyl-CoA (products of a running cycle — feedback inhibition)
- Citrate itself exits the mitochondria during fatty acid synthesis (citrate shuttle)
STEP 2 — Citrate → Isocitrate
- Enzyme: Aconitase
- Reaction type: Dehydration then rehydration (isomerisation via cis-aconitate intermediate)
- Reversible
- Converts the tertiary alcohol of citrate to the secondary alcohol of isocitrate (needed for the next oxidation step)
- Inhibited by fluorocitrate (metabolite of fluoroacetate/fluoroacetamide poisoning)
- Aconitase contains an iron-sulfur [4Fe-4S] cluster — inhibited by nitric oxide and in iron deficiency
STEP 3 — Isocitrate → α-Ketoglutarate
- Enzyme: Isocitrate dehydrogenase (IDH)
- Reaction type: Oxidative decarboxylation
- Irreversible
- 1 NADH + 1 CO₂ produced
- This is often cited as the rate-limiting step of the TCA cycle
- Activated by: ADP, Ca²⁺, NAD⁺, isocitrate (substrate)
- Inhibited by: ATP, NADH (energy excess signals)
- Note: IDH2 mutation (gain of function) is found in gliomas and AML — produces 2-hydroxyglutarate (an oncometabolite)
STEP 4 — α-Ketoglutarate → Succinyl-CoA
- Enzyme: α-Ketoglutarate dehydrogenase complex (α-KGDH)
- Reaction type: Oxidative decarboxylation
- Irreversible
- 1 NADH + 1 CO₂ produced
- Cofactors: TPP (Thiamine B1), Lipoic acid, CoA, FAD, NAD⁺ — identical set as pyruvate dehydrogenase
- Inhibited by: Succinyl-CoA (product), NADH, ATP, arsenic compounds
- This is the second major thiamine-dependent step in oxidative metabolism (PDH is the first)
- Inhibited by arsenite (binds lipoic acid — same mechanism as in PDH inhibition)
STEP 5 — Succinyl-CoA → Succinate
- Enzyme: Succinyl-CoA synthetase (succinate thiokinase)
- Reaction type: Substrate-level phosphorylation
- Reversible
- 1 GTP produced (in most tissues; ATP in heart and liver — same energetic value)
- This is the only step in the TCA cycle that produces a high-energy phosphate directly (substrate-level phosphorylation — like glycolysis)
- Succinyl-CoA is also a key intermediate: it is the entry point for propionyl-CoA (from odd-chain fatty acid oxidation and certain amino acids), and it is the starting point for haem synthesis (combines with glycine via ALA synthase)
STEP 6 — Succinate → Fumarate
- Enzyme: Succinate dehydrogenase (SDH / Complex II of the ETC)
- Reaction type: Oxidation
- Reversible
- 1 FADH₂ produced (not NADH — the oxidation potential of succinate → fumarate is insufficient to reduce NAD⁺)
- Unique: SDH is embedded in the inner mitochondrial membrane — it is the only TCA cycle enzyme that is also part of the electron transport chain
- Inhibited by: Malonate (competitive inhibitor — structural analogue of succinate, the classic example of competitive inhibition in biochemistry)
- SDH mutations cause hereditary paraganglioma/phaeochromocytoma syndromes (SDHB, SDHC, SDHD genes)
STEP 7 — Fumarate → Malate
- Enzyme: Fumarase (fumarate hydratase)
- Reaction type: Hydration (adds H₂O)
- Reversible
- Fumarase mutations cause fumaric aciduria (autosomal recessive — encephalopathy, renal cysts)
- Fumarate is also produced from the urea cycle (argininosuccinate lyase step) and from purine synthesis — connecting TCA to nitrogen metabolism
STEP 8 — Malate → Oxaloacetate
- Enzyme: Malate dehydrogenase (MDH)
- Reaction type: Oxidation
- Reversible
- 1 NADH produced
- Regenerates oxaloacetate, completing the cycle
- MDH also exists as a cytoplasmic isoform (MDH1) — participates in the malate-aspartate shuttle (transfers NADH reducing equivalents into the mitochondria)
- This reaction is thermodynamically unfavourable (ΔG° is positive) — it proceeds because OAA is immediately consumed by citrate synthase in Step 1 (product removal drives the reaction)
Complete Energy Yield Per Turn of the TCA Cycle
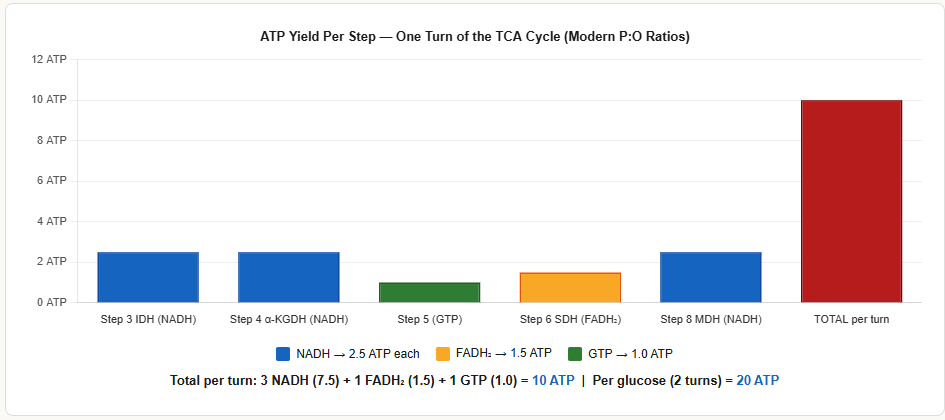
| Step | Product | ATP Equivalent (Modern) |
|---|---|---|
| Step 3 (IDH) | 1 NADH | 2.5 ATP |
| Step 4 (α-KGDH) | 1 NADH | 2.5 ATP |
| Step 5 (Succinyl-CoA synthetase) | 1 GTP | 1.0 ATP |
| Step 6 (SDH) | 1 FADH₂ | 1.5 ATP |
| Step 8 (MDH) | 1 NADH | 2.5 ATP |
| TOTAL per turn | 3 NADH + 1 FADH₂ + 1 GTP | 10 ATP |
Per glucose (2 turns): 6 NADH + 2 FADH₂ + 2 GTP = 20 ATP from the TCA cycle alone
(Plus 2 NADH from pyruvate dehydrogenase × 2 = additional 5 ATP)
The Pyruvate Dehydrogenase Complex (PDH) — The Gateway to the TCA Cycle
PDH is not formally part of the TCA cycle, but it is the essential entry point that converts pyruvate to Acetyl-CoA. You cannot understand TCA regulation without it.
Reaction: Pyruvate + CoA + NAD⁺ → Acetyl-CoA + CO₂ + NADH
Cofactors (same 5 as α-KGDH):
- TPP (Thiamine B1) — decarboxylation
- Lipoic acid — carries the acetyl group
- CoA — accepts the acetyl group
- FAD — regenerates lipoic acid
- NAD⁺ — final electron acceptor
Regulation of PDH:
| Activators | Inhibitors |
|---|---|
| Pyruvate (substrate) | Acetyl-CoA (product) |
| ADP | NADH (product) |
| Ca²⁺ (exercise) | ATP |
| Insulin (activates PDH phosphatase) | Glucagon/Epinephrine (activate PDH kinase → phosphorylation → inactive) |
| CoA | Succinyl-CoA |
PDH Deficiency:
- X-linked (most common form) or autosomal recessive
- Pyruvate cannot be converted to Acetyl-CoA → pyruvate accumulates → converted to lactate → lactic acidosis
- Presents in neonates with neurological deterioration, Leigh syndrome
- Ketogenic diet provides Acetyl-CoA directly from fat, bypassing the PDH block
⚙️ Section 2 — Regulation of the TCA Cycle
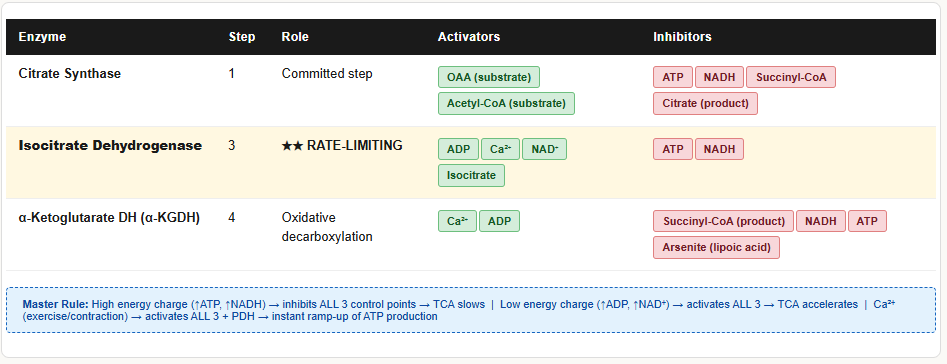
The TCA cycle is regulated at 3 irreversible steps (Steps 1, 3, 4) and at the PDH gateway. The overall principle is simple: high energy charge (high ATP, high NADH) slows the cycle; low energy charge (high ADP, high NAD⁺) speeds it up.
The Three Main Control Points
Control Point 1: Citrate Synthase (Step 1)
| Inhibitors | Notes |
|---|---|
| ATP | High energy — no need to generate more |
| NADH | Reduction equivalents already abundant |
| Succinyl-CoA | Downstream product — feedback inhibition |
| Citrate (product) | Product inhibition |
No known allosteric activators — rate is controlled primarily by substrate availability (OAA, Acetyl-CoA).
Control Point 2: Isocitrate Dehydrogenase (Step 3) — Rate-Limiting
| Activators | Inhibitors |
|---|---|
| ADP (low energy signal) | ATP |
| Ca²⁺ (muscle contraction) | NADH |
| NAD⁺ |
Control Point 3: α-Ketoglutarate Dehydrogenase (Step 4)
| Activators | Inhibitors |
|---|---|
| Ca²⁺ | Succinyl-CoA (product) |
| ADP | NADH |
| ATP | |
| Arsenic compounds |
The Role of Calcium — The Exercise Signal
Ca²⁺ is a critical activator of three enzymes simultaneously: PDH phosphatase (activates PDH), isocitrate dehydrogenase, and α-ketoglutarate dehydrogenase. When a muscle contracts, Ca²⁺ floods into the cytoplasm — the same signal that triggers contraction simultaneously signals the mitochondria to ramp up ATP production. This is elegant, coordinated physiology.
Energy Charge Summary
| Condition | Signal | Effect on TCA |
|---|---|---|
| ATP high, ADP low (rest) | High energy charge | Citrate synthase, IDH, α-KGDH all inhibited → cycle slows |
| ADP high, ATP low (exercise/work) | Low energy charge | All 3 enzymes activated → cycle accelerates |
| NADH high | Electron carriers saturated | All 3 enzymes inhibited (ETC cannot accept more) |
| Ca²⁺ high (exercise) | Contraction signal | PDH, IDH, α-KGDH all activated |
🔄 Section 3 — Anaplerosis: Feeding the Cycle and Draining It
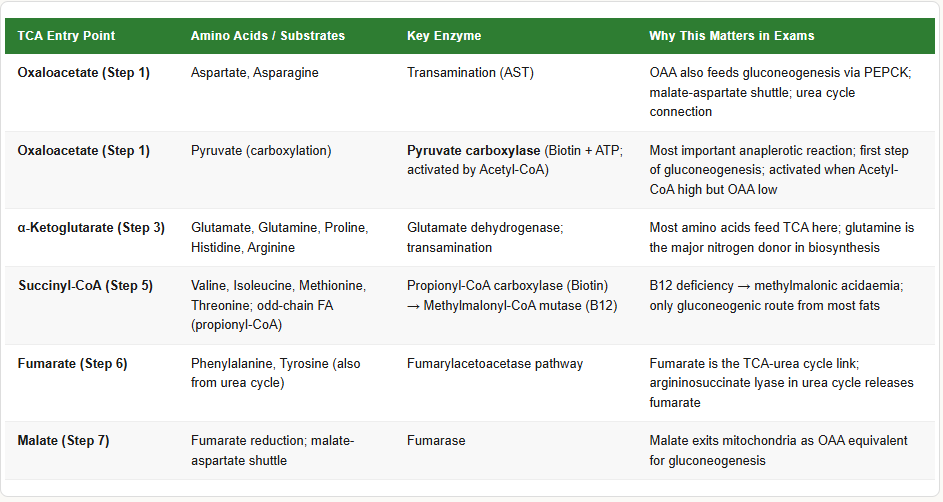
What Is Anaplerosis?
The TCA cycle intermediates do not just go around in a circle — they are constantly being drained (removed for biosynthesis) and replenished (via anaplerotic reactions). If intermediates are not replenished, the cycle stalls even if Acetyl-CoA is abundant.
Key Anaplerotic Reactions (replenishing OAA):
| Substrate | Entry Point | Enzyme | Notes |
|---|---|---|---|
| Pyruvate | Oxaloacetate | Pyruvate carboxylase | Rate-limiting step of gluconeogenesis; activated by acetyl-CoA; requires Biotin |
| Glutamate / Glutamine | α-Ketoglutarate | Glutamate dehydrogenase | Most important amino acid entry point |
| Aspartate | Oxaloacetate | Transamination (AST) | Also connects urea cycle to TCA |
| Propionyl-CoA (odd-chain FA) | Succinyl-CoA | Propionyl-CoA carboxylase → methylmalonyl-CoA mutase (B12) | Only gluconeogenic substrate from FA |
| Phenylalanine / Tyrosine | Fumarate | Fumarylacetoacetase pathway | |
| Isoleucine / Methionine / Valine | Succinyl-CoA | Multiple steps |
Key Cataplerotic (draining) Reactions:
| Intermediate Removed | For What Purpose |
|---|---|
| Citrate | Exits to cytoplasm → fatty acid synthesis (citrate shuttle) |
| α-Ketoglutarate | → Glutamate synthesis; → transamination reactions |
| Succinyl-CoA | → Haem synthesis (with glycine via ALA synthase) |
| Oxaloacetate | → Gluconeogenesis (via PEPCK); → aspartate synthesis |
| Malate | → Gluconeogenesis; → exits mitochondria (malate-aspartate shuttle) |
Pyruvate Carboxylase — The Most Important Anaplerotic Enzyme
- Reaction: Pyruvate + CO₂ + ATP → Oxaloacetate
- Cofactor: Biotin
- Location: Mitochondrial matrix
- Activated by: Acetyl-CoA (when Acetyl-CoA is high but OAA is low, pyruvate carboxylase is activated to replenish OAA and keep the cycle running)
- This is also the first step of gluconeogenesis — OAA → PEPCK → PEP
🏥 Section 4 — Clinical Connections
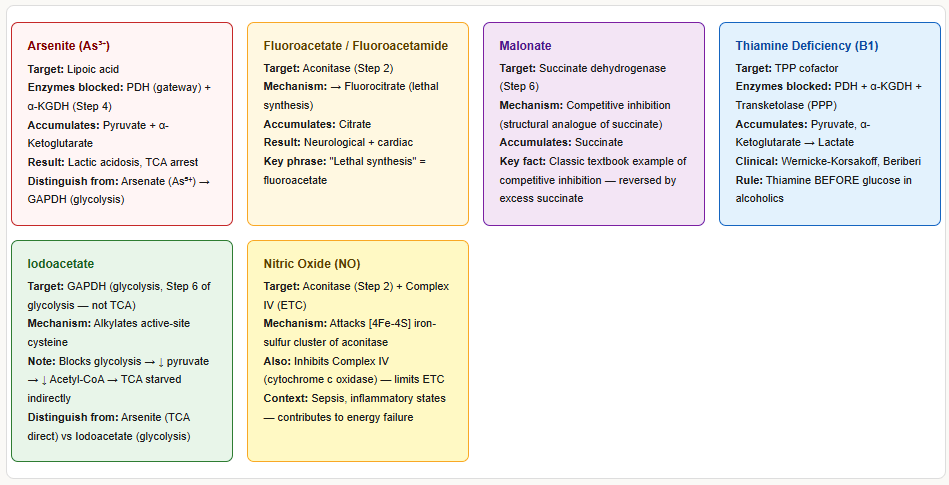
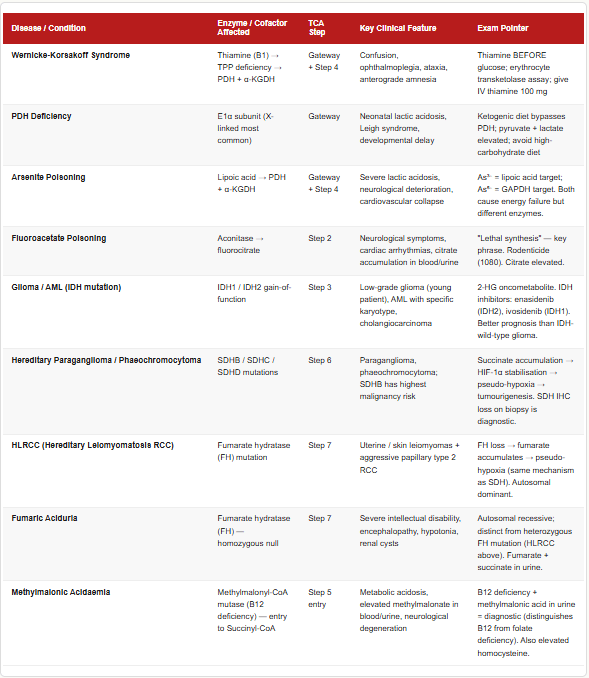
Thiamine (Vitamin B1) Deficiency
Thiamine deficiency affects two TCA-linked enzymes: PDH (gateway) and α-KGDH (Step 4). It also affects transketolase in the pentose phosphate pathway.
Result: Pyruvate cannot enter TCA → accumulates → converted to lactate → lactic acidosis. α-Ketoglutarate cannot be processed → accumulates.
Clinical presentations:
- Wernicke encephalopathy: Confusion, ophthalmoplegia, ataxia (triad) — acute, reversible with thiamine
- Korsakoff syndrome: Anterograde amnesia, confabulation — chronic, often irreversible
- Wet beriberi: High-output cardiac failure, oedema (peripheral vasodilation from ATP deficit)
- Dry beriberi: Peripheral neuropathy (sensorimotor)
Diagnosis: Erythrocyte transketolase activity (before and after TPP addition) — the TPP effect (>25% increase = deficiency)
Key exam fact: Give thiamine BEFORE glucose in any alcoholic patient — giving glucose first depletes the last reserves of thiamine and can precipitate acute Wernicke encephalopathy.
Arsenite Poisoning
- Arsenite (As³⁺) binds and inactivates lipoic acid — the cofactor shared by PDH and α-KGDH
- Blocks both enzymes simultaneously → pyruvate and α-ketoglutarate accumulate → lactic acidosis + TCA cycle arrest
- Clinically: severe lactic acidosis, neurological deterioration, cardiovascular collapse
- Contrast with arsenate (As⁵⁺): acts at GAPDH in glycolysis (mimics phosphate) — different mechanism
Fluoroacetate / Fluoroacetamide Poisoning (Rodenticide)
- Fluoroacetate is metabolised to fluoroacetyl-CoA → condenses with OAA → fluorocitrate
- Fluorocitrate is a powerful inhibitor of aconitase (Step 2)
- TCA cycle is blocked at citrate → citrate accumulates
- Called the “lethal synthesis” — the body synthesises the actual toxin from an innocuous precursor
- Clinical: severe neurological symptoms, cardiac arrhythmias, death
Isocitrate Dehydrogenase (IDH) Mutations — Cancer Connection
- IDH1 (cytoplasmic) and IDH2 (mitochondrial) gain-of-function mutations
- Mutant IDH converts α-ketoglutarate → 2-hydroxyglutarate (2-HG), an oncometabolite
- 2-HG inhibits α-ketoglutarate-dependent dioxygenases → blocks DNA demethylation → epigenetic dysregulation → cancer
- Found in: Low-grade glioma (most common), glioblastoma, AML (acute myeloid leukaemia), cholangiocarcinoma
- Diagnostic: IDH mutation testing by PCR/sequencing; serum 2-HG elevated
- Treatment: IDH inhibitors — enasidenib (IDH2), ivosidenib (IDH1)
Succinate Dehydrogenase (SDH) Mutations — Hereditary Tumour Syndromes
- SDHB, SDHC, SDHD, SDHAF2 mutations → SDH complex dysfunction → succinate accumulates
- Accumulated succinate inhibits prolyl hydroxylase → HIF-1α is not degraded → pseudo-hypoxia → tumour growth
- Clinical: Hereditary paraganglioma-phaeochromocytoma syndrome
- SDHB mutation: highest malignancy risk (paraganglioma, phaeochromocytoma, renal cell carcinoma, GI stromal tumour)
- Autosomal dominant with variable penetrance
Fumarase (Fumarate Hydratase) Deficiency
- Autosomal recessive
- Fumaric aciduria → severe intellectual disability, failure to thrive, renal cysts
- Fumarate hydratase is also a tumour suppressor: FH mutations (heterozygous) cause hereditary leiomyomatosis and renal cell carcinoma (HLRCC)
Pyruvate Dehydrogenase (PDH) Deficiency
- Most common: E1α subunit deficiency (X-linked)
- Pyruvate cannot be converted to Acetyl-CoA → pyruvate → lactate → lactic acidosis
- Presents: neonatal lactic acidosis, Leigh syndrome (subacute necrotising encephalopathy), developmental delay
- Treatment: ketogenic diet (bypasses PDH by providing Acetyl-CoA directly from fat)
🔗 Section 5 — TCA Cycle Connections to Other Pathways
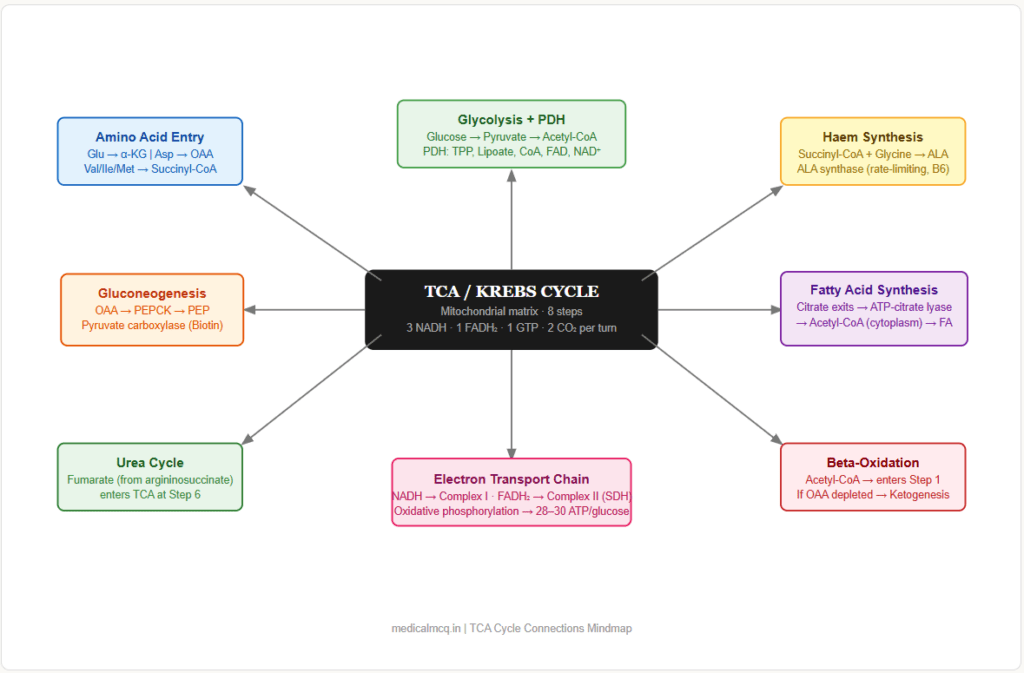
→ Glycolysis — Pyruvate (end product of glycolysis) → PDH → Acetyl-CoA → enters TCA at Step 1. The TCA cycle is the downstream continuation of glycolysis under aerobic conditions. When TCA is inhibited (e.g., thiamine deficiency), pyruvate backs up → lactate formation → lactic acidosis.
→ Fatty Acid Oxidation — Beta-oxidation produces Acetyl-CoA, which enters the TCA cycle at Step 1. Also produces NADH and FADH₂ that compete for the ETC. When TCA cycle is overwhelmed (OAA depleted in fasting), excess Acetyl-CoA → ketogenesis.
→ Amino Acid Catabolism — Multiple amino acids enter the TCA cycle at different points (anaplerosis). Glutamate → α-KG (Step 3 entry). Aspartate → OAA. Isoleucine, methionine, valine → Succinyl-CoA (Step 5 entry).
→ Gluconeogenesis — OAA (from the TCA cycle) is the direct precursor for gluconeogenesis via PEPCK (OAA → PEP). The malate-aspartate shuttle transfers OAA indirectly. Propionyl-CoA → succinyl-CoA → malate → OAA → gluconeogenesis (the only gluconeogenic pathway from fat).
→ Haem Synthesis — Succinyl-CoA (Step 5 product) + Glycine → ALA (δ-aminolaevulinic acid) via ALA synthase — the first and rate-limiting step of haem synthesis. Pyridoxal phosphate (B6) is the cofactor.
→ Amino Acid Synthesis — α-Ketoglutarate → Glutamate (by glutamate dehydrogenase or transamination) → Glutamine, Proline, Arginine, Histidine. OAA → Aspartate (by transamination) → Asparagine, nucleotide synthesis.
→ Lipid Synthesis — Citrate exits the mitochondria → ATP-citrate lyase → Acetyl-CoA (cytoplasm) → fatty acid synthesis, cholesterol synthesis. The TCA cycle therefore feeds both energy production AND fat building.
→ Electron Transport Chain — NADH and FADH₂ produced in the TCA cycle are the electron donors for Complexes I and II of the ETC respectively. The TCA cycle cannot run without a functioning ETC to reoxidise NADH → NAD⁺.
🎯 High-Yield Exam Facts
These are the exact facts that appear repeatedly across NEET PG, USMLE, AIIMS and FMGE papers.
🔴 Isocitrate dehydrogenase is the rate-limiting enzyme of the TCA cycle The most commonly tested single fact about TCA regulation. Activated by ADP and Ca²⁺; inhibited by ATP and NADH.
🔴 The TCA cycle produces 3 NADH + 1 FADH₂ + 1 GTP + 2 CO₂ per turn = 10 ATP Per glucose = 2 turns = 20 ATP from TCA. Add PDH contribution (2 NADH = 5 ATP) = 25 ATP from TCA+PDH.
🔴 Thiamine (B1) is required by PDH, α-KGDH, and transketolase — ALL three cause lactic acidosis if deficient The shared cofactor is TPP (thiamine pyrophosphate). Deficiency → Wernicke-Korsakoff, beriberi, lactic acidosis.
🔴 Succinate dehydrogenase (Step 6) is the only TCA enzyme embedded in the inner mitochondrial membrane — it is also Complex II of the ETC This dual identity is tested repeatedly. All other TCA enzymes are soluble in the matrix (except aconitase).
🔴 Fluoroacetate → fluorocitrate → inhibits aconitase (Step 2) — “lethal synthesis” The key phrase “lethal synthesis” is a direct exam answer. Citrate accumulates.
🔴 Arsenite binds lipoic acid → inhibits PDH and α-KGDH (Steps 4 + gateway) Different from arsenate (inhibits GAPDH in glycolysis). Arsenite = lipoic acid target; arsenate = phosphate mimic.
🟠 The only substrate-level phosphorylation in the TCA cycle is at Step 5 (succinyl-CoA synthetase) → produces GTP (or ATP) This is the TCA equivalent of ATP generation without the ETC. Tested alongside glycolytic substrate-level phosphorylation.
🟠 Malonate is a competitive inhibitor of succinate dehydrogenase — the textbook example of competitive inhibition Malonate is structurally similar to succinate; competes for the active site. Effect is overcome by adding excess succinate.
🟠 Pyruvate carboxylase (anaplerotic) is activated by Acetyl-CoA When Acetyl-CoA is high but OAA is low (fasting state), Acetyl-CoA signals pyruvate carboxylase to replenish OAA — ensuring the cycle can run. Requires Biotin and ATP.
🟠 Succinyl-CoA is the entry point for haem synthesis — connects TCA to haematology ALA synthase uses Succinyl-CoA + Glycine → ALA (requires pyridoxal phosphate / B6). ALA synthase is the rate-limiting enzyme of haem synthesis.
🟠 IDH1/IDH2 gain-of-function mutations → 2-hydroxyglutarate (2-HG) → glioma, AML The oncometabolite produced by mutant IDH is 2-HG, not α-ketoglutarate. IDH inhibitors: enasidenib (IDH2), ivosidenib (IDH1).
🟡 Give thiamine BEFORE glucose in alcoholics — NOT after Giving glucose first forces the last TPP reserves to be used for PDH → precipitates acute Wernicke encephalopathy. This is one of the highest-yield clinical management facts linked to TCA.
🟡 Fumarate is produced by BOTH the TCA cycle AND the urea cycle Fumarate exits the urea cycle (from argininosuccinate lyase) and enters the TCA cycle. This links nitrogen disposal to energy metabolism and is a favourite integration question.
🟡 OAA (oxaloacetate) → malate → exits to cytoplasm → used in gluconeogenesis OAA cannot cross the inner mitochondrial membrane directly. It is converted to malate or aspartate for transport, then reconverted. This matters for gluconeogenesis questions.
🟡 α-Ketoglutarate dehydrogenase has the same 5 cofactors as PDH: TPP, Lipoic acid, CoA, FAD, NAD⁺ The mnemonic “TLC for FaN” (TPP, Lipoic acid, CoA, FAD, NAD⁺) applies to both. Both are inhibited by arsenite.
🧠 Mnemonics & Memory Tricks
“Citrate Is Krebs’ Starting Substrate For Making Oxaloacetate” → Helps you remember: All 8 TCA intermediates in order
C = Citrate (Step 1) I = Isocitrate (Step 2) K = α-Ketoglutarate (Step 3) S = Succinyl-CoA (Step 4) S = Succinate (Step 5) F = Fumarate (Step 6) M = Malate (Step 7) O = Oxaloacetate (Step 8)
💡 Pro tip: This is the single most important TCA mnemonic. Memorise it first. Every step question, every “which comes after” question, every enzyme question — they all become trivial once you have this sequence locked in.
“TLC for FaN” — Cofactors of PDH and α-KGDH → Helps you remember all 5 shared cofactors
T = TPP (Thiamine pyrophosphate) L = Lipoic acid C = CoA (Coenzyme A) F = FAD N = NAD⁺
💡 Pro tip: When a question says “which vitamin is required by both PDH and the TCA cycle enzyme α-KGDH?” — the answer is Thiamine (B1). When it asks “which cofactor is the target of arsenite?” — lipoic acid (the L in TLC).
“3 Irreversible Steps = Steps 1, 3, 4 — The CSI of TCA” → Helps you remember the 3 irreversible (regulated) steps
C = Citrate synthase (Step 1) S = iSocitrate dehydrogenase (Step 3) — rate-limiting I = α-ketoglutarate dehydrogenase (α-KGDH) (Step 4)
💡 Pro tip: These are also the 3 steps inhibited by high energy charge (ATP, NADH). Any exam question about “which steps of the TCA cycle are regulated?” = CSI.
Energy Shortcut — “Count the Dehydrogenations” → For any TCA question asking about yield
Rule: Every time you see “dehydrogenase” in the TCA cycle, it produces one electron carrier. Count the dehydrogenases:
- Steps 3, 4, 8 use NAD⁺ → 3 NADH
- Step 6 uses FAD → 1 FADH₂
- Step 5 is the only substrate-level phosphorylation → 1 GTP → Total = 3 NADH + 1 FADH₂ + 1 GTP per turn
💡 Pro tip: You can reconstruct the entire energy yield table on the spot with this rule. No memorisation of numbers required — just count the dehydrogenases.
⚡ Exam Shortcuts — Answer TCA Questions in Under 30 Seconds
These are pattern-recognition tricks for the most frequently tested TCA question types.
Shortcut 1 — “Which intermediate accumulates when X is inhibited?” Method: Find the blocked step on your mnemonic sequence. The intermediate BEFORE that step accumulates.
- Fluoroacetate blocks Step 2 (Aconitase) → Citrate accumulates (Step 1 product)
- Arsenite blocks Step 4 (α-KGDH) → α-Ketoglutarate accumulates (Step 3 product)
- Malonate blocks Step 6 (SDH) → Succinate accumulates (Step 5 product)
Shortcut 2 — “Where does amino acid X enter the TCA cycle?” Method: Look at the carbon skeleton of the amino acid.
- Glutamate / Glutamine / Proline / Histidine / Arginine → α-Ketoglutarate (Step 3)
- Aspartate / Asparagine → Oxaloacetate (Step 8)
- Valine / Isoleucine / Methionine / Threonine → Succinyl-CoA (Step 4)
- Phenylalanine / Tyrosine → Fumarate (Step 6) + Acetoacetate
- Tryptophan → multiple points
Shortcut 3 — “How many ATP from X turns of TCA?” Method: 10 ATP per turn × number of turns
- 1 Acetyl-CoA = 1 turn = 10 ATP
- 1 Glucose = 2 Acetyl-CoA = 2 turns = 20 ATP (TCA only)
- 1 Palmitate (C16) = 8 Acetyl-CoA = 8 turns = 80 ATP (TCA component only)
Shortcut 4 — “Which TCA enzyme is deficient in [clinical scenario]?” Method: Match the cofactor clue.
- Confusion + ophthalmoplegia + lactic acidosis in alcoholic → Thiamine deficiency → PDH + α-KGDH (Steps 4 + gateway)
- “Lethal synthesis” / rodenticide / citrate accumulation → Fluoroacetate → Aconitase (Step 2)
- Paraganglioma / phaeochromocytoma + family history → SDH mutation (Step 6)
- Glioma + young patient / AML → IDH mutation (Step 3)
Shortcut 5 — “Which step produces FADH₂ not NADH?” Answer: Always Step 6 (Succinate → Fumarate, Succinate dehydrogenase). In the entire TCA cycle, only one step uses FAD instead of NAD⁺.
⚠️ Common Mistakes Students Make on This Topic
❌ Mistake: “The TCA cycle is separate from the electron transport chain” ✅ Reality: Succinate dehydrogenase (Step 6) is both a TCA enzyme and Complex II of the ETC — embedded in the inner mitochondrial membrane. The two systems are directly coupled. 📝 How this gets tested: “Which TCA cycle enzyme is also a component of the respiratory chain?” — the answer is SDH / Complex II. Students who think all TCA enzymes are matrix-soluble choose the wrong option.
❌ Mistake: “NADH and FADH₂ yield the same amount of ATP” ✅ Reality: NADH yields 2.5 ATP; FADH₂ yields 1.5 ATP (modern P:O ratios). FADH₂ enters the ETC at Complex II (bypasses Complex I), so it yields less. 📝 How this gets tested: ATP yield calculation questions. Using old ratios (3 ATP for NADH, 2 for FADH₂) gives slightly different totals — NEET PG questions may still use older values (38 ATP), so know both systems.
❌ Mistake: “Arsenite and arsenate have the same mechanism” ✅ Reality: Arsenite (As³⁺) binds lipoic acid → inhibits PDH and α-KGDH. Arsenate (As⁵⁺) mimics phosphate → inhibits GAPDH in glycolysis. Two different arsenic species, two different enzymes, two different mechanisms. 📝 How this gets tested: “A patient with arsenic poisoning shows lactic acidosis — which form of arsenic and which enzyme?” — the valency (3+ vs 5+) determines the mechanism.
❌ Mistake: “The TCA cycle only runs in the fed state” ✅ Reality: The TCA cycle runs continuously in all aerobic cells. It INCREASES in the fasted state as fatty acid oxidation floods it with Acetyl-CoA. What changes in fasting is the SOURCE of Acetyl-CoA (fat instead of carbohydrates) and the FATE of OAA (pulled into gluconeogenesis). 📝 How this gets tested: Integration questions asking about metabolic flux in fasted liver cells — the TCA cycle is very active even in fasting, but OAA availability limits it and forces Acetyl-CoA into ketogenesis.
❌ Mistake: “CO₂ released in the TCA cycle comes from the Acetyl-CoA carbons” ✅ Reality: The 2 CO₂ released per turn come from oxaloacetate carbons (at Steps 3 and 4) — NOT from the incoming Acetyl-CoA carbons. The carbons from Acetyl-CoA are first incorporated into citrate and only lost as CO₂ in subsequent turns of the cycle. 📝 How this gets tested: Isotope labelling questions — “If ¹⁴C-labelled Acetyl-CoA enters the cycle, when does ¹⁴CO₂ appear?” The answer is: NOT in the first turn — in subsequent turns.
📝 5 Practice MCQs — Test Yourself Now
Q1: Which enzyme catalyses the rate-limiting step of the TCA cycle?
- A. Citrate synthase
- B. α-Ketoglutarate dehydrogenase
- C. Isocitrate dehydrogenase
- D. Malate dehydrogenase
✅ Answer: C. Isocitrate dehydrogenase
Why correct: Isocitrate dehydrogenase (IDH) at Step 3 is the rate-limiting, most regulated enzyme of the TCA cycle. It is allosterically activated by ADP and Ca²⁺ and inhibited by ATP and NADH — making it the primary throttle for the entire cycle’s speed.
Why A is wrong: Citrate synthase is the first and committed step and is regulated, but the rate-limiting designation belongs to IDH. Citrate synthase is controlled mainly by substrate availability and product inhibition. Why B is wrong: α-KGDH (Step 4) is an important regulated step sharing the same inhibitors as IDH, but IDH is accepted as the rate-limiting enzyme in most standard biochemistry texts used for NEET PG and USMLE. Why D is wrong: Malate dehydrogenase (Step 8) is a reversible enzyme with no major regulatory role — it is thermodynamically unfavourable in isolation and is driven by mass action, not allosteric regulation.
Exam tip: “Rate-limiting enzyme of TCA cycle” = Isocitrate dehydrogenase. This is one of the five single most important rate-limiting enzyme facts in biochemistry (along with PFK-1, ACC, CAT-I, and HMG-CoA reductase).
Q2: A 40-year-old chronic alcoholic is brought to the emergency department with confusion, lateral gaze palsy, and difficulty walking. Blood glucose is 110 mg/dL but lactate is elevated at 4.8 mmol/L. The treating physician gives intravenous dextrose immediately. What is the most dangerous consequence of this decision?
- A. Hyperglycaemia worsening cerebral oedema
- B. Rapid depletion of residual thiamine, precipitating acute Wernicke encephalopathy
- C. Lactic acidosis worsening due to increased pyruvate production
- D. Inhibition of gluconeogenesis by high insulin release
✅ Answer: B. Rapid depletion of residual thiamine, precipitating acute Wernicke encephalopathy
Why correct: This patient has Wernicke encephalopathy from thiamine deficiency (the triad: confusion, ophthalmoplegia, ataxia). Giving glucose forces PDH to process the resulting pyruvate, consuming the last traces of TPP. This abruptly destroys whatever residual function PDH had and precipitates fulminant Wernicke encephalopathy. The correct protocol is IV thiamine FIRST, then glucose.
Why A is wrong: At a glucose of 110 mg/dL, hyperglycaemia is not a concern. Cerebral oedema from hyperglycaemia would require much higher glucose levels. Why C is wrong: While lactic acidosis may worsen slightly, this is not the most dangerous consequence — it would happen regardless. The dangerous consequence is the acute neurological deterioration from thiamine depletion. Why D is wrong: Gluconeogenesis inhibition by insulin at physiological glucose levels is not dangerous and is a normal physiological response, not a complication.
Exam tip: “Alcoholic + confusion + ophthalmoplegia + ataxia → Give thiamine BEFORE glucose” is one of the most directly tested clinical management facts linked to TCA cycle biochemistry. It appears in both NEET PG and USMLE clinical vignettes.
Q3: Fluoroacetate is used as a rodenticide in agriculture. A farm worker is poisoned and presents with severe neurological symptoms. Laboratory analysis of his blood would most likely show elevation of which metabolite?
- A. α-Ketoglutarate
- B. Succinate
- C. Citrate
- D. Pyruvate
✅ Answer: C. Citrate
Why correct: Fluoroacetate is metabolised to fluoroacetyl-CoA, which condenses with oxaloacetate to form fluorocitrate. Fluorocitrate is a potent inhibitor of aconitase (Step 2 — Citrate → Isocitrate). Blocking Step 2 means citrate cannot be converted to isocitrate, so citrate accumulates. This phenomenon is called “lethal synthesis” because the body builds the active toxin from a harmless precursor.
Why A is wrong: α-Ketoglutarate accumulates when Step 4 (α-KGDH) is blocked — caused by arsenite or thiamine deficiency, not fluoroacetate. Why B is wrong: Succinate accumulates when Step 6 (SDH) is blocked — caused by malonate poisoning. Why D is wrong: Pyruvate accumulates when PDH is blocked (thiamine deficiency, arsenite, PDH deficiency) — not specifically from fluoroacetate poisoning.
Exam tip: The phrase “lethal synthesis” in any question stem = fluoroacetate = fluorocitrate = aconitase inhibition = citrate accumulation. This word sequence is the answer key.
Q4: A 28-year-old man is diagnosed with a grade 2 glioma. Tumour sequencing reveals an IDH1 R132H point mutation. What is the direct biochemical consequence of this mutation?
- A. Increased conversion of isocitrate to α-ketoglutarate and excess NADH
- B. Conversion of α-ketoglutarate to 2-hydroxyglutarate by gain-of-function enzymatic activity
- C. Loss of IDH1 function leading to citrate accumulation
- D. Increased OAA production leading to accelerated gluconeogenesis
✅ Answer: B. Conversion of α-ketoglutarate to 2-hydroxyglutarate by gain-of-function enzymatic activity
Why correct: IDH1 R132H is a gain-of-function mutation (not a loss-of-function). The mutant enzyme gains a new neomorphic activity: it reduces α-ketoglutarate to 2-hydroxyglutarate (2-HG) using NADPH. 2-HG is an oncometabolite that competitively inhibits α-ketoglutarate-dependent dioxygenases (including TET2 DNA demethylases and histone demethylases) → epigenetic reprogramming → gliomagenesis.
Why A is wrong: This describes normal IDH1 function. The mutant enzyme loses its normal isocitrate → α-KG activity and gains the new 2-HG-producing activity instead. Why C is wrong: Citrate accumulation occurs with aconitase inhibition (fluoroacetate), not IDH1 mutation. Why D is wrong: IDH1 does not produce OAA. The mutation does not accelerate gluconeogenesis.
Exam tip: IDH mutations in cancer = gain of function = 2-HG production (not loss of normal function). IDH inhibitors: ivosidenib (IDH1), enasidenib (IDH2). Found in: glioma, AML, cholangiocarcinoma.
Q5: A metabolic biochemistry researcher blocks the TCA cycle at Step 6 using malonate in an isolated mitochondria preparation. Which of the following best describes the effect on upstream intermediates and the electron transport chain?
- A. α-Ketoglutarate and succinyl-CoA accumulate; Complex I is overloaded
- B. Succinate accumulates; Complex II receives no substrate; upstream NADH production continues
- C. Citrate accumulates; the entire ETC shuts down immediately
- D. Fumarate accumulates; Complex I and III are directly inhibited
✅ Answer: B. Succinate accumulates; Complex II receives no substrate; upstream NADH production continues
Why correct: Malonate competitively inhibits succinate dehydrogenase (SDH = Complex II). Step 6 is blocked → succinate accumulates (the substrate for Step 6). Steps 1–5 continue to run, producing NADH (at Steps 3, 4, 8) normally — NADH can still donate electrons to Complex I. However, Complex II (SDH) receives no succinate → FADH₂ is not produced from this step → the FADH₂ contribution to the ETC is lost. The cycle itself does not stop entirely — only the SDH step is impaired.
Why A is wrong: α-Ketoglutarate and succinyl-CoA accumulate when Steps 4 is blocked (α-KGDH inhibition). Malonate blocks Step 6, not Step 4. Complex I is not “overloaded” — NADH production continues normally but the ETC may actually slow because overall cycle flux decreases. Why C is wrong: Citrate accumulates when Step 2 (aconitase) is blocked by fluorocitrate. Malonate does not affect aconitase. The ETC does not shut down immediately — NADH-linked complexes continue. Why D is wrong: Fumarate is the product of Step 6 — it would be reduced (not accumulated) if SDH is blocked. Complex I and III are not directly inhibited by malonate.
Exam tip: Malonate = competitive SDH inhibitor = succinate accumulates = FADH₂ from SDH lost = classic competitive inhibition example. Reversible by adding excess succinate — this is how competitive inhibition is demonstrated experimentally.
📚 References
📖 Harper’s Illustrated Biochemistry — Rodwell et al. | Chapter 16: The Citric Acid Cycle: The Central Pathway of Carbohydrate, Lipid & Amino Acid Metabolism
📖 Lippincott’s Illustrated Reviews: Biochemistry — Ferrier | Chapter 9: Tricarboxylic Acid Cycle
📖 Lehninger Principles of Biochemistry — Nelson & Cox | Chapter 16: The Citric Acid Cycle
📖 Stryer’s Biochemistry — Berg, Tymoczko & Stryer | Chapter 17: The Citric Acid Cycle
📖 Vasudevan & Sreekumari’s Textbook of Biochemistry — Vasudevan | Chapter 7: Biological Oxidation and TCA Cycle
🚀 Keep Practising — You Are Not Done Yet
Reading this article gives you the foundation. But the TCA cycle questions that appear in NEET PG, USMLE, and AIIMS are almost always clinical scenarios — not pure recall. A patient presents with something. You need to trace the biochemistry back to an enzyme, a cofactor, or a metabolite.
The only way to build that clinical reasoning speed is deliberate practice under timed conditions.
medicalmcq.in has free Biochemistry MCQs covering every pathway in this article — TCA cycle, PDH, anaplerosis, clinical disorders — all with detailed explanations that build your mechanistic thinking.
Do 10–15 questions immediately after reading. That converts reading into memory. Then revisit in 48 hours for spaced repetition. That converts memory into exam performance.