What You Will Learn in This Article
- Name all 4 unique enzymes of gluconeogenesis and explain exactly why each is needed
- List all major gluconeogenic substrates and trace each one’s entry point into the pathway
- Explain why fatty acids (with one exception) cannot be used for gluconeogenesis
- State the exact ATP cost of synthesising one glucose molecule from pyruvate
- Describe hormonal regulation — how glucagon, cortisol, and insulin control the pathway
- Identify the key allosteric regulators including the role of Fructose-2,6-bisphosphate
- Connect gluconeogenesis to the Cori cycle, glucose-alanine cycle, and clinical disorders
- Recognise the clinical basis of hypoglycaemia, metformin toxicity, and von Gierke disease
📖 Introduction: Why This Topic Matters in Exams
It is 3 AM. A patient who has been fasting for 18 hours comes to the emergency department confused and diaphoretic. Blood glucose is 38 mg/dL. The question is not just “give glucose” — the question is why the patient became hypoglycaemic in the first place, and that requires understanding what the body normally does during prolonged fasting to prevent exactly this scenario. That mechanism is gluconeogenesis — the synthesis of glucose from non-carbohydrate precursors. When it fails, patients die.
Gluconeogenesis is tested across every format in NEET PG and USMLE Step 1. Pure recall questions (“which enzyme is unique to gluconeogenesis?”), mechanistic questions (“why can’t even-chain fatty acids contribute to net gluconeogenesis?”), regulatory questions (“what does glucagon do to fructose-2,6-bisphosphate?”), and full clinical scenarios (“a patient on metformin develops lactic acidosis — what is the mechanism?”). It is one of the most interconnected topics in biochemistry, which is exactly why it appears so often.
This article covers the complete pathway from all substrates to glucose, the 4 unique bypass enzymes that make it work, full regulatory biochemistry at both the allosteric and hormonal levels, the clinical disorders, and 5 exam-style MCQs to test whether you have genuinely understood the material.
🔬 Section 1 — The Big Picture: What Gluconeogenesis Is and Where It Happens
Definition and Purpose
Gluconeogenesis is the metabolic pathway that synthesises glucose from non-carbohydrate precursors. It is the primary mechanism by which the body maintains blood glucose during:
- Prolonged fasting (beyond 8–12 hours, once glycogen stores are depleted)
- Starvation
- Intense exercise (especially in liver and kidney)
- Low-carbohydrate states
- Diabetes mellitus (particularly Type 2 — unregulated gluconeogenesis is a major cause of fasting hyperglycaemia)
Where It Occurs
Primary site: Liver — responsible for ~90% of gluconeogenesis during fasting. Liver is unique in expressing glucose-6-phosphatase, the final enzyme that releases free glucose into the blood.
Secondary site: Renal cortex — becomes increasingly important during prolonged starvation (can contribute up to 40% of glucose production during extended fasting). Renal gluconeogenesis uses glutamine as the preferred substrate.
Intestinal epithelium — minor contribution, uses glutamine; relevant during post-absorptive state.
NOT in muscle: Muscle lacks glucose-6-phosphatase and cannot release free glucose into the blood. Muscle can perform gluconeogenic reactions internally but cannot complete the pathway to free glucose.
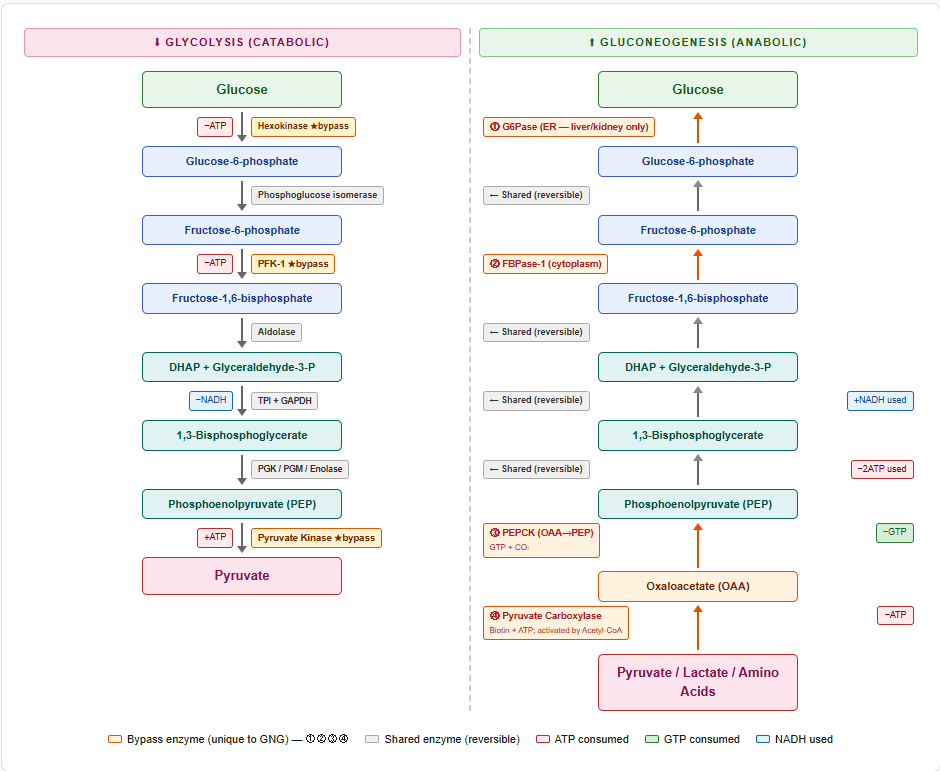
The Core Concept: Reversing Glycolysis — But Not Completely
Gluconeogenesis is broadly the reverse of glycolysis, sharing 7 of the 10 steps. However, the 3 irreversible steps of glycolysis create thermodynamic barriers that cannot simply be run backwards. Three unique bypass reactions overcome these barriers:
| Irreversible Glycolytic Step | Enzyme | Gluconeogenic Bypass Enzyme(s) |
|---|---|---|
| Pyruvate kinase (PEP → Pyruvate) | Pyruvate kinase | Pyruvate carboxylase + PEPCK (two-step bypass) |
| PFK-1 (Fructose-6-P → Fructose-1,6-bisP) | PFK-1 | Fructose-1,6-bisphosphatase (FBPase-1) |
| Hexokinase / Glucokinase (Glucose → Glucose-6-P) | Hexokinase | Glucose-6-phosphatase |
These 4 enzymes (pyruvate carboxylase, PEPCK, FBPase-1, glucose-6-phosphatase) are unique to gluconeogenesis and are the most-tested facts about this pathway.
🔬 Section 2 — Gluconeogenic Substrates: What Goes In
The Major Substrates
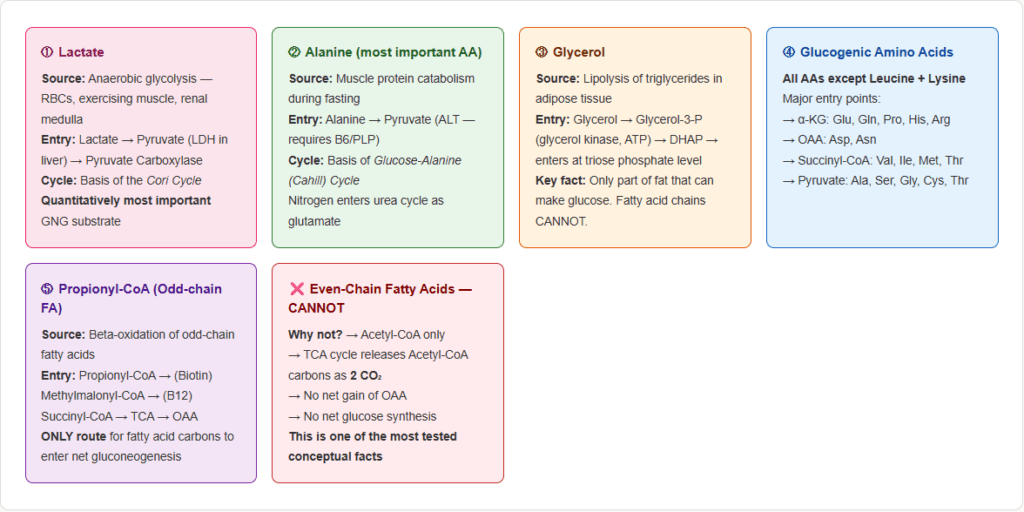
1. Lactate — the most important gluconeogenic substrate quantitatively
- Source: Anaerobic glycolysis in RBCs, exercising muscle, renal medulla
- Entry point: Converted to pyruvate by lactate dehydrogenase (LDH) in the liver
- This is the basis of the Cori cycle (see Section 5)
- Lactate → Pyruvate → (via pyruvate carboxylase + PEPCK) → PEP → … → Glucose
2. Amino acids (glucogenic amino acids)
All amino acids except leucine and lysine are at least partly glucogenic. The most important:
| Amino Acid | Entry Point | Key Notes |
|---|---|---|
| Alanine | Pyruvate | Most important glucogenic amino acid; the glucose-alanine cycle |
| Glutamine | α-Ketoglutarate | Major substrate in kidney; also important in intestine |
| Glutamate | α-Ketoglutarate | Via transamination |
| Aspartate | OAA | Direct transamination |
| Serine | Pyruvate | Via serine dehydratase |
| Glycine | Pyruvate | Via serine intermediate |
| Threonine | Pyruvate or Succinyl-CoA | |
| Methionine / Valine / Isoleucine | Succinyl-CoA | Odd-carbon intermediates → TCA → OAA → GNG |
| Histidine, Proline, Arginine | Glutamate → α-KG | |
| Phenylalanine / Tyrosine | Fumarate | Also ketogenic (Acetoacetate) |
Purely ketogenic amino acids (CANNOT contribute to gluconeogenesis): Leucine and Lysine only.
Both are converted exclusively to Acetyl-CoA/Acetoacetate — and Acetyl-CoA cannot be converted to net glucose in mammals (the carbon is lost as CO₂ in TCA before OAA is regenerated).
3. Glycerol
- Source: Lipolysis of triglycerides in adipose tissue releases glycerol
- Entry point: Glycerol → Glycerol-3-phosphate (glycerol kinase) → DHAP (glycerol-3-P dehydrogenase) → enters gluconeogenesis at the triose phosphate level
- Only the glycerol backbone of fats can contribute to glucose synthesis — the fatty acid chains cannot (in mammals, for even-chain fatty acids)
4. Propionate / Propionyl-CoA (from odd-chain fatty acids)
- Odd-chain fatty acids yield propionyl-CoA as the final product of beta-oxidation
- Propionyl-CoA → (propionyl-CoA carboxylase, Biotin) → Methylmalonyl-CoA → (methylmalonyl-CoA mutase, B12) → Succinyl-CoA → TCA → OAA → gluconeogenesis
- This is the only route by which fatty acid carbons can contribute to net glucose synthesis
Why Even-Chain Fatty Acids Cannot Support Net Gluconeogenesis
This is one of the highest-yield conceptual questions on this topic. Here is the reasoning:
- Even-chain fatty acids are broken down entirely to Acetyl-CoA (C2 units)
- Acetyl-CoA enters the TCA cycle by condensing with OAA → Citrate
- In each turn, the 2 carbons from Acetyl-CoA are released as 2 CO₂ — before OAA is regenerated
- The OAA that emerges at the end of the cycle is derived from the original OAA, not from Acetyl-CoA
- Therefore, there is no net gain of OAA from Acetyl-CoA — the carbon atoms of Acetyl-CoA are entirely lost as CO₂
- Without net new OAA, there is no net new glucose
The simple version: Acetyl-CoA carbons → CO₂ in TCA. Nothing is gained. No glucose is made.
The exception: Odd-chain fatty acids → propionyl-CoA → succinyl-CoA → OAA (net gain) → glucose. This IS gluconeogenic.
🔬 Section 3 — The 4 Unique Enzymes: The Bypass Reactions in Detail
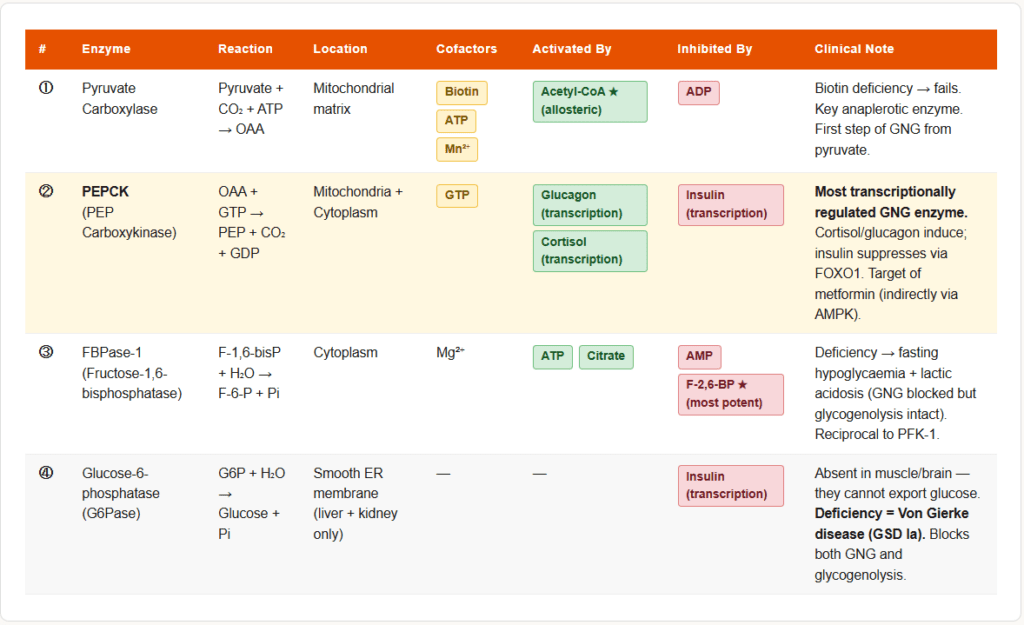
Bypass 1: Pyruvate → Phosphoenolpyruvate (PEP) — A Two-Step Process
This bypass replaces the irreversible pyruvate kinase reaction. It requires two separate enzymes and costs energy.
Step 1a: Pyruvate → Oxaloacetate
- Enzyme: Pyruvate carboxylase
- Location: Mitochondrial matrix
- Cofactor: Biotin (covalently attached; carries CO₂), ATP, Mn²⁺
- Reaction: Pyruvate + CO₂ + ATP → Oxaloacetate + ADP + Pi
- Activation: Allosterically activated by Acetyl-CoA (the key signal: when fatty acid oxidation is active and Acetyl-CoA is abundant, gluconeogenesis is simultaneously stimulated)
- Regulation: Pyruvate carboxylase is also the first committed step of gluconeogenesis when the substrate is pyruvate or lactate
Step 1b: Oxaloacetate → PEP
- Enzyme: Phosphoenolpyruvate carboxykinase (PEPCK)
- Location: Both mitochondria and cytoplasm (the isoforms differ by species and tissue)
- Cofactor: GTP (the phosphate donor)
- Reaction: Oxaloacetate + GTP → PEP + CO₂ + GDP
- Important: OAA cannot cross the inner mitochondrial membrane directly. It must first be converted to either malate (by mitochondrial MDH) or aspartate (by transamination), which then cross the membrane and are reconverted to OAA in the cytoplasm.
- Transcription: PEPCK gene is induced by glucagon and cortisol (via cAMP and glucocorticoid response elements) and suppressed by insulin. This is the most transcriptionally regulated enzyme in gluconeogenesis.
- Combined cost of Bypass 1: 2 ATP equivalents (1 ATP for pyruvate carboxylase + 1 GTP for PEPCK)
Bypass 2: Fructose-1,6-bisphosphate → Fructose-6-phosphate
- Enzyme: Fructose-1,6-bisphosphatase (FBPase-1)
- Location: Cytoplasm
- Reaction: Fructose-1,6-bisphosphate + H₂O → Fructose-6-phosphate + Pi (simple hydrolysis)
- Inhibited by: AMP (low energy signal), Fructose-2,6-bisphosphate (F-2,6-BP — the most potent inhibitor)
- Activated by: ATP, citrate
- This is the exact reciprocal of PFK-1 in glycolysis. When FBPase-1 is active, PFK-1 is inhibited, and vice versa — the same molecules (F-2,6-BP, AMP, ATP) regulate both enzymes in opposite directions.
Bypass 3: Glucose-6-phosphate → Glucose
- Enzyme: Glucose-6-phosphatase (G6Pase)
- Location: Smooth ER membrane of liver and kidney cells (NOT present in muscle or brain)
- Reaction: Glucose-6-phosphate + H₂O → Glucose + Pi (simple hydrolysis)
- This enzyme is what gives the liver the unique ability to export free glucose into the bloodstream
- Deficiency: Von Gierke disease (Glycogen storage disease Type Ia) — G6Pase deficiency means neither glycogenolysis NOR gluconeogenesis can release free glucose → severe fasting hypoglycaemia + hepatomegaly + lactic acidosis + hyperlipidaemia
⚙️ Section 4 — Regulation of Gluconeogenesis
The Master Principle: Reciprocal Regulation with Glycolysis
Gluconeogenesis and glycolysis are reciprocally regulated — when one is active, the other is suppressed. This prevents futile cycling. The same key molecules regulate both pathways in opposite directions.
Allosteric Regulation
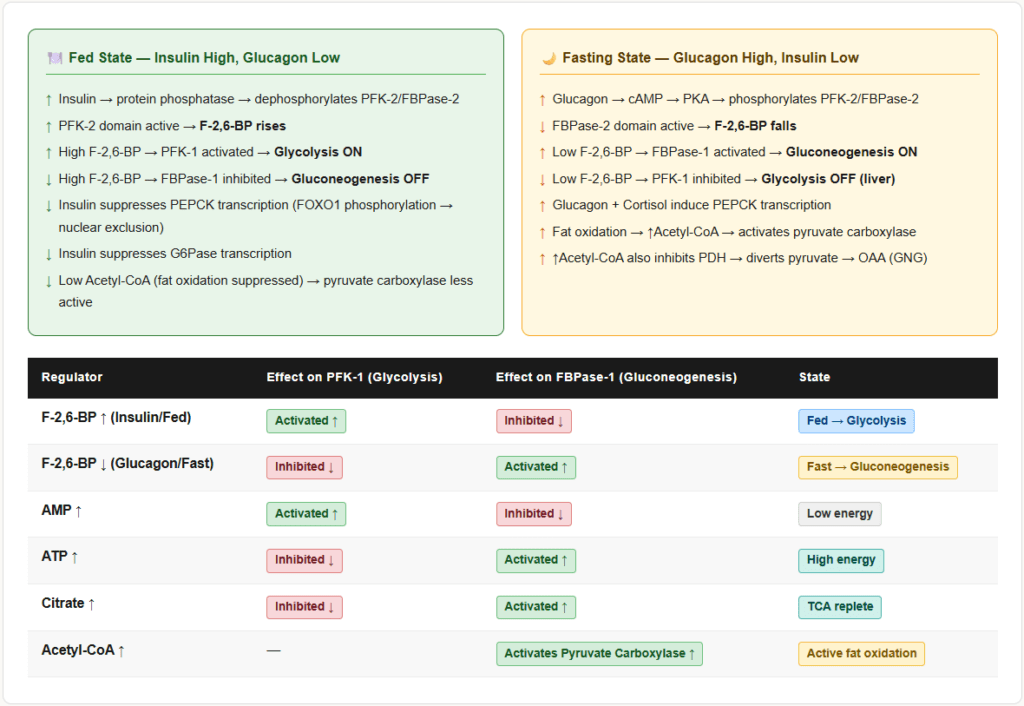
Fructose-2,6-bisphosphate (F-2,6-BP) — The Most Important Allosteric Regulator
F-2,6-BP is a regulatory molecule produced by the bifunctional enzyme PFK-2/FBPase-2:
- In the fed state: Insulin activates protein phosphatase → dephosphorylates PFK-2/FBPase-2 → PFK-2 domain is active → F-2,6-BP rises → PFK-1 activated (glycolysis ON) + FBPase-1 inhibited (gluconeogenesis OFF)
- In the fasted state: Glucagon → cAMP → PKA → phosphorylates PFK-2/FBPase-2 → FBPase-2 domain is active → F-2,6-BP falls → FBPase-1 activated (gluconeogenesis ON) + PFK-1 inhibited (glycolysis OFF)
This single molecule — F-2,6-BP — is the most important allosteric switch between glycolysis and gluconeogenesis.
AMP — The Energy Sensor
- High AMP (low energy state, e.g., exercise, fasting): Inhibits FBPase-1 (suppresses gluconeogenesis) AND activates PFK-1 (stimulates glycolysis) AND activates AMPK
- Wait — this seems counterintuitive. If fasting causes high AMP, why does gluconeogenesis still run?
- Answer: During fasting, the dominant signal is low F-2,6-BP (from glucagon), not just AMP. The hormonal signal via glucagon overrides the AMP effect on FBPase-1 at the transcriptional level. PEPCK and G6Pase are upregulated transcriptionally, compensating.
Acetyl-CoA — Activating Pyruvate Carboxylase
- In fasting: Fatty acid oxidation generates abundant Acetyl-CoA in the liver
- Acetyl-CoA allosterically activates pyruvate carboxylase → pushes pyruvate toward OAA → feeds gluconeogenesis
- Acetyl-CoA also inhibits pyruvate dehydrogenase (PDH) → prevents pyruvate from being consumed by TCA, redirecting it to gluconeogenesis
- These two effects together mean that active fat oxidation simultaneously fuels gluconeogenesis. Elegant design.
ATP, Citrate — Activating FBPase-1
- High ATP activates FBPase-1 (gluconeogenesis) and inhibits PFK-1 (glycolysis)
- Citrate activates FBPase-1 and inhibits PFK-1
Summary Table: Allosteric Control at the Key Steps
| Molecule | Effect on Glycolysis (PFK-1) | Effect on Gluconeogenesis (FBPase-1) |
|---|---|---|
| F-2,6-BP ↑ (insulin/fed) | ↑ Activated | ↓ Inhibited |
| F-2,6-BP ↓ (glucagon/fasted) | ↓ Inhibited | ↑ Activated |
| AMP ↑ | ↑ Activated | ↓ Inhibited |
| ATP ↑ | ↓ Inhibited | ↑ Activated |
| Citrate ↑ | ↓ Inhibited | ↑ Activated |
| Acetyl-CoA ↑ | — | ↑ Activates pyruvate carboxylase |
Hormonal Regulation
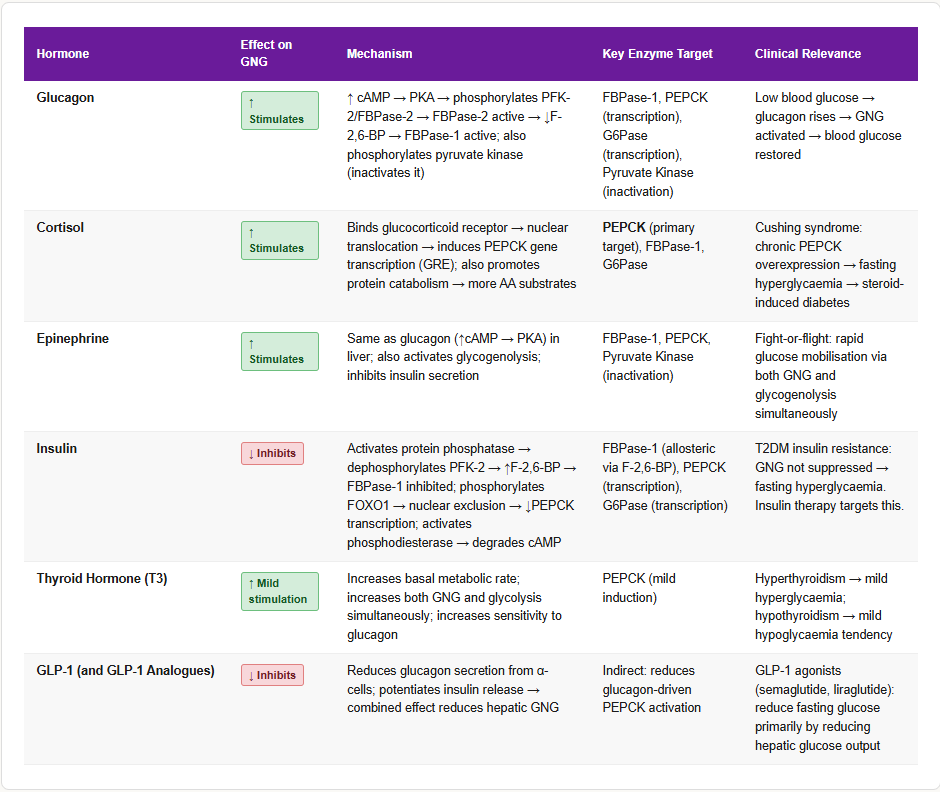
Glucagon — The Primary Fasting Hormone
- Released by pancreatic α-cells when blood glucose falls
- Activates adenylyl cyclase → ↑ cAMP → activates PKA
- PKA phosphorylates PFK-2/FBPase-2 → FBPase-2 active → ↓ F-2,6-BP → FBPase-1 active → gluconeogenesis ON
- PKA also phosphorylates pyruvate kinase → inactivates it → prevents PEP from being converted back to pyruvate (removes futile cycling)
- Transcriptionally induces: PEPCK, G6Pase, FBPase-1 (sustained gluconeogenesis)
Cortisol — The Stress Hormone
- Induces transcription of PEPCK (the most cortisol-responsive GNG enzyme)
- Promotes protein catabolism → releases amino acids → more gluconeogenic substrate
- Induces expression of gluconeogenic enzymes over hours to days
- Works synergistically with glucagon
- This is why Cushing syndrome causes hyperglycaemia and why patients on corticosteroids develop steroid-induced diabetes
Insulin — The Fed-State Inhibitor
- Activates protein phosphatase → dephosphorylates PFK-2/FBPase-2 → PFK-2 active → ↑ F-2,6-BP → FBPase-1 inhibited
- Activates phosphodiesterase → degrades cAMP → opposes glucagon signalling
- Transcriptionally suppresses: PEPCK, G6Pase (via FOXO1 phosphorylation → nuclear exclusion)
- Activates pyruvate kinase (promotes glycolysis)
Epinephrine — Emergency State
- Same mechanism as glucagon (↑ cAMP → PKA) in liver
- In the emergency state (fight or flight), gluconeogenesis is maximally stimulated alongside glycogenolysis
Thyroid Hormone (T3)
- Increases basal metabolic rate → increases gluconeogenesis AND glycolysis simultaneously
- Net effect is usually hyperglycaemia in hyperthyroidism
🧮 Section 5 — The Energy Cost of Gluconeogenesis
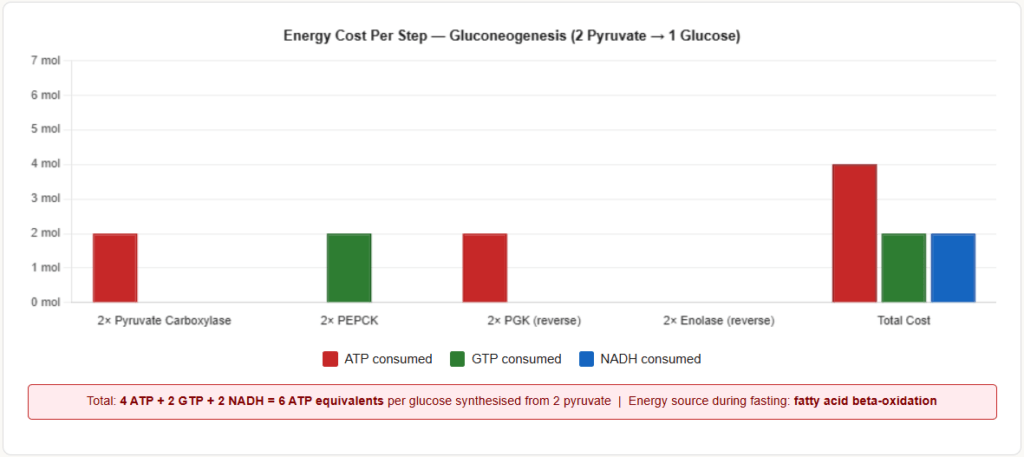
Synthesising glucose from pyruvate is energetically expensive — this is why the body does not do it frivolously.
Stoichiometry of gluconeogenesis from 2 pyruvate → 1 glucose:
| Step | Energy Cost |
|---|---|
| 2× Pyruvate carboxylase (2 pyruvate → 2 OAA) | 2 ATP |
| 2× PEPCK (2 OAA → 2 PEP) | 2 GTP |
| 2× Phosphoglycerate kinase (reversal) | 2 ATP |
| 2× Enolase reversal (phosphoglycerate mutase) | — |
| Total | 4 ATP + 2 GTP = 6 ATP equivalents |
| NADH consumed (glyceraldehyde-3-P dehydrogenase, running in reverse) | 2 NADH |
Full equation:
2 Pyruvate + 4 ATP + 2 GTP + 2 NADH → Glucose + 4 ADP + 2 GDP + 6 Pi + 2 NAD⁺
Compare: Glycolysis produces 2 ATP net per glucose. Gluconeogenesis costs 6 ATP equivalents to reverse the process. The body accepts this cost only when the alternative (no blood glucose) is worse.
Source of ATP for gluconeogenesis during fasting: Fatty acid beta-oxidation in the liver provides the ATP and NADH needed to drive gluconeogenesis. This is another reason why fat oxidation and gluconeogenesis are activated together in fasting.
🔄 Section 6 — The Cori Cycle and Glucose-Alanine Cycle
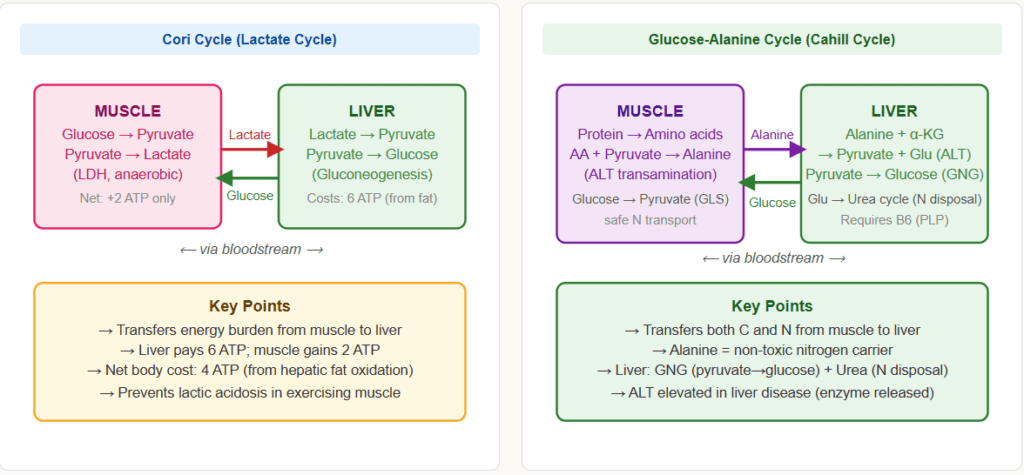
The Cori Cycle (Lactate Cycle)
The Cori cycle is the interorgan metabolic cycle between exercising muscle and the liver:
- Muscle: Under anaerobic conditions (intense exercise), muscle performs glycolysis → pyruvate → lactate (LDH) → lactate exported to blood
- Liver: Lactate taken up → converted to pyruvate (LDH) → enters gluconeogenesis → glucose exported to blood
- Muscle: Receives glucose from blood → glycolysis again
Net effect: The liver uses ATP (from fat oxidation) to convert muscle-derived lactate back to glucose, which the muscle can use again. The liver effectively subsidises anaerobic muscle metabolism.
Key exam fact: The Cori cycle transfers the energy burden from muscle to liver. Muscle gets glucose without paying the ATP cost; liver pays the 6 ATP cost of gluconeogenesis using energy from fat oxidation.
Named after: Carl and Gerty Cori — both Nobel laureates (1947). Gerty Cori was the first American woman to win a Nobel Prize in science.
The Glucose-Alanine Cycle (Cahill Cycle)
Similar to the Cori cycle but uses alanine instead of lactate:
- Muscle: Amino acids released from protein catabolism (during fasting/exercise) → transaminated with pyruvate → alanine exported to blood
- Liver: Alanine taken up → transaminated back to pyruvate → enters gluconeogenesis → glucose exported
- The nitrogen from alanine enters the urea cycle in the liver
Why alanine? Alanine is a non-toxic amino acid carrier. Muscle cannot dispose of nitrogen from amino acid catabolism directly — it packages nitrogen as alanine for safe transport to the liver.
Net effect: The glucose-alanine cycle transfers both carbon (as glucose precursors) and nitrogen (as alanine’s amino group) from muscle to liver during fasting and exercise.
🏥 Section 7 — Clinical Connections
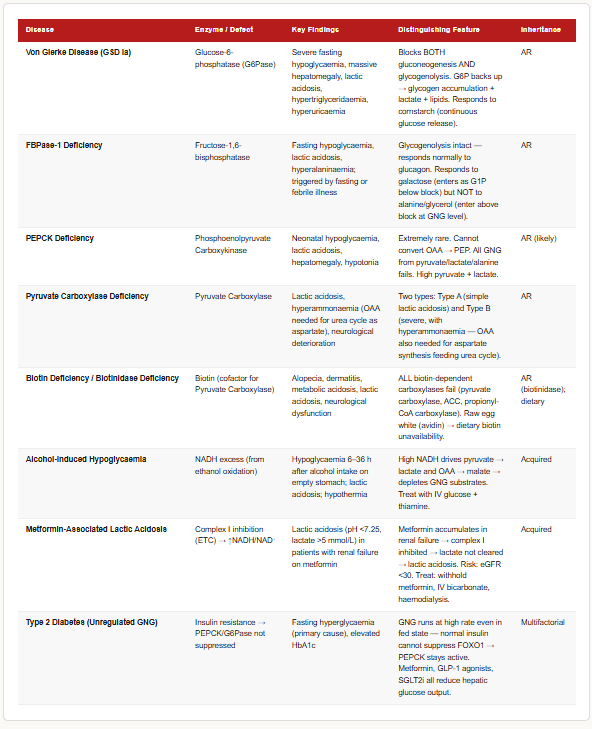
Von Gierke Disease (Glycogen Storage Disease Type Ia)
- Defect: Glucose-6-phosphatase deficiency (the final enzyme of gluconeogenesis AND glycogenolysis)
- Inheritance: Autosomal recessive
- Consequence: Neither glycogenolysis NOR gluconeogenesis can release free glucose → severe fasting hypoglycaemia
- Metabolic consequences: Glucose-6-phosphate accumulates → diverted to glycolysis → pyruvate → lactate → lactic acidosis | Diverted to pentose phosphate pathway → excess NADPH | G6P → glycolysis → Acetyl-CoA → hyperlipidaemia | Hyperuricaemia (from ATP consumption)
- Clinical features: Hepatomegaly (glycogen accumulation), fasting hypoglycaemia, lactic acidosis, hyperlipidaemia, hyperuricaemia (gout), doll-like facies
- Treatment: Frequent feeds, cornstarch supplementation, nocturnal glucose/nasogastric feeding; liver transplant in severe cases
Fructose-1,6-bisphosphatase Deficiency
- Defect: FBPase-1 deficiency — the second bypass enzyme
- Inheritance: Autosomal recessive
- Consequence: Gluconeogenesis is blocked at the FBPase-1 step → cannot convert triose phosphates to glucose → hypoglycaemia when glycogen stores are depleted
- Clinical features: Fasting hypoglycaemia, lactic acidosis, hyperalaninemia (substrates back up), episodes triggered by fasting or febrile illness
- Distinguishes from Von Gierke: Responds to fructose/glycerol/alanine loading (which enter above the block) but NOT to galactose (which enters as G6P, below the block)
Metformin and Lactic Acidosis
- Mechanism of Metformin: Inhibits Complex I of the mitochondrial electron transport chain in the liver → reduces NADH reoxidation → mitochondrial NADH/NAD⁺ ratio rises → pyruvate → lactate (LDH is driven toward lactate when NADH is high) → simultaneously, the fall in mitochondrial ATP production inhibits gluconeogenesis (less ATP for pyruvate carboxylase and PEPCK)
- Metformin also activates AMPK → AMPK phosphorylates and inactivates gluconeogenic enzymes (including TORC2, a transcriptional coactivator of PEPCK)
- Net effect: Reduced hepatic gluconeogenesis → lower fasting blood glucose — the primary therapeutic mechanism in T2DM
- Lactic acidosis risk: In patients with renal impairment, liver disease, or hypoxia, the backup of lactate (which the liver cannot clear because its own gluconeogenesis is impaired) causes life-threatening lactic acidosis
- Contraindications: eGFR <30 mL/min, liver disease, heart failure, conditions causing hypoxia
PEPCK Deficiency
- Extremely rare — only a handful of cases reported
- Neonatal hypoglycaemia, lactic acidosis, hepatomegaly
- PEPCK is so central to gluconeogenesis that its absence is essentially incompatible with normal neonatal metabolism after glycogen stores are depleted
Biotin Deficiency
- Pyruvate carboxylase requires biotin as a cofactor
- Biotin deficiency (from raw egg white ingestion — avidin binds biotin, making it unavailable) or genetic biotinidase/holocarboxylase synthetase deficiency
- Impairs pyruvate carboxylase → cannot convert pyruvate to OAA → gluconeogenesis from lactate/pyruvate/alanine fails → hypoglycaemia + lactic acidosis
- Also affects propionyl-CoA carboxylase and methylcrotonyl-CoA carboxylase → organic acidaemias
- Features: alopecia, dermatitis, neurological dysfunction, metabolic acidosis
Alcohol-Induced Hypoglycaemia
- Ethanol is oxidised by alcohol dehydrogenase (ADH) → Acetaldehyde → (ALDH) → Acetate
- Both steps use NAD⁺ → produce NADH → liver NADH/NAD⁺ ratio rises dramatically
- High NADH drives: Pyruvate → Lactate (LDH) | OAA → Malate (MDH)
- Both pyruvate AND OAA — the two key gluconeogenic substrates — are depleted
- Gluconeogenesis fails → hypoglycaemia, especially in fasting alcoholics
- Simultaneously: High NADH impairs fatty acid oxidation → hepatic fat accumulation (alcoholic fatty liver)
- Clinical: Alcoholic hypoglycaemia occurs 6–36 hours after binge drinking on an empty stomach
Diabetes Mellitus Type 2 — Unregulated Gluconeogenesis
- In T2DM, there is insulin resistance in the liver → insulin’s suppression of PEPCK and G6Pase is lost
- Gluconeogenesis runs at high rates even in the fed state when it should be suppressed
- This is the primary cause of fasting hyperglycaemia in T2DM — not glucose absorption, but unregulated hepatic glucose production
- Metformin, GLP-1 agonists, and SGLT2 inhibitors all reduce hepatic glucose output to varying degrees
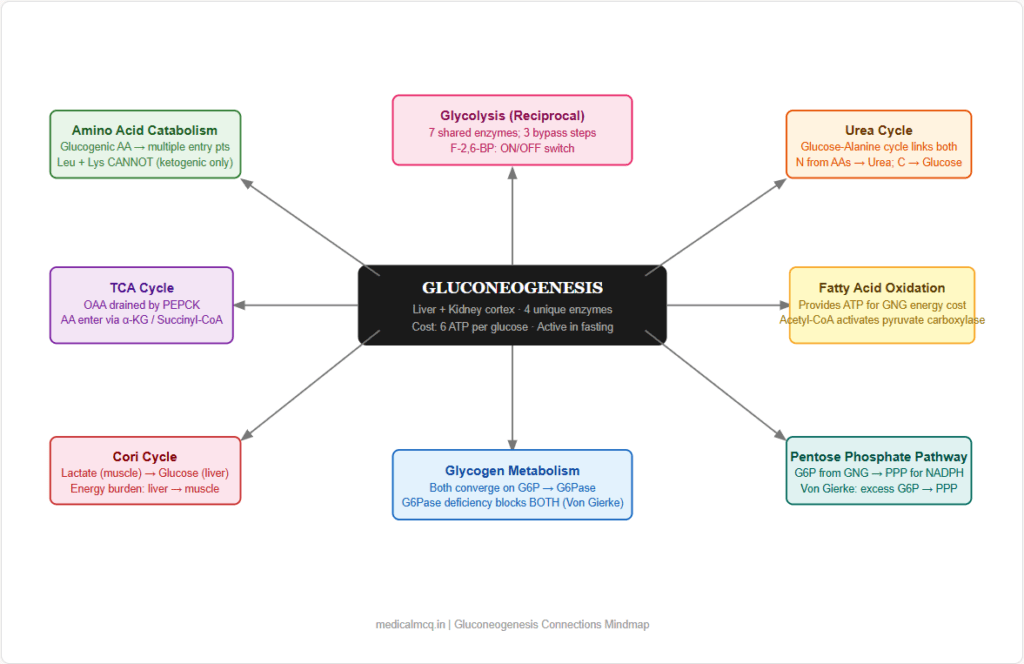
🔗 Section 8 — Connections to Other Pathways
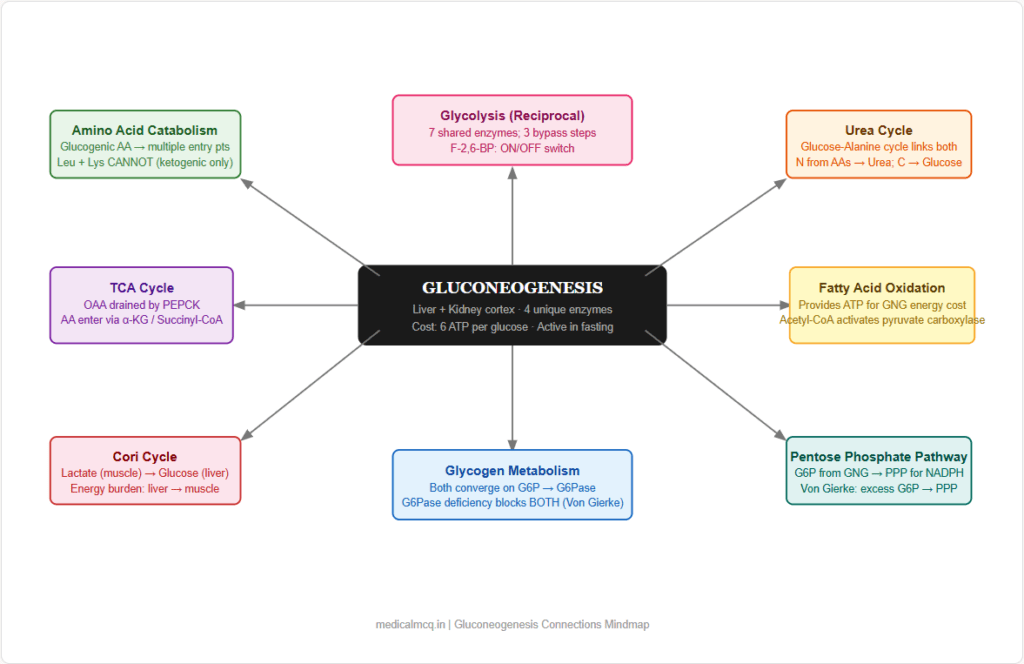
→ Glycolysis — The mirror image. Seven steps are shared and run in reverse. The 3 irreversible glycolytic steps are each bypassed by a unique gluconeogenic enzyme. Reciprocal regulation by F-2,6-BP, AMP, ATP, citrate ensures the two pathways do not run simultaneously in the same direction.
→ TCA Cycle — OAA is the key connecting intermediate. TCA cycle intermediates (OAA, malate, fumarate, succinyl-CoA, α-ketoglutarate) feed into gluconeogenesis via anaplerosis. PEPCK converts OAA to PEP, draining the TCA cycle when gluconeogenesis is active (cataplerosis).
→ Fatty Acid Oxidation — Beta-oxidation provides the ATP, NADH, and Acetyl-CoA that fuel and activate gluconeogenesis. Acetyl-CoA activates pyruvate carboxylase; beta-oxidation produces the ATP needed for the energy-expensive steps. Fatty acid oxidation and gluconeogenesis are always co-activated in fasting.
→ Amino Acid Catabolism — Glucogenic amino acids provide carbon skeletons at multiple entry points. Muscle protein catabolism in fasting/starvation is the body’s last resort for gluconeogenic substrate when glycogen is gone and glycerol is insufficient.
→ Urea Cycle — The nitrogen from glucogenic amino acids must be disposed of. Amino acid transamination → aspartate → enters urea cycle. The glucose-alanine cycle explicitly couples gluconeogenesis (in liver) with urea synthesis (in liver) and muscle protein catabolism.
→ Glycogen Metabolism — Both glycogenolysis and gluconeogenesis are regulated by glucagon/insulin and both converge on glucose-6-phosphate → glucose (via G6Pase). Von Gierke disease (G6Pase deficiency) simultaneously blocks both pathways. In the early fasting state, glycogenolysis dominates; after ~12–16 hours, gluconeogenesis takes over.
→ Pentose Phosphate Pathway — Glucose-6-phosphate produced by gluconeogenesis can be diverted to the PPP instead of being released as glucose. This provides NADPH and ribose-5-phosphate for biosynthesis.
🎯 High-Yield Exam Facts
These are the exact facts that appear repeatedly across NEET PG, USMLE, AIIMS and FMGE papers.
🔴 The 4 unique enzymes of gluconeogenesis: Pyruvate carboxylase, PEPCK, FBPase-1, Glucose-6-phosphatase Every other enzyme is shared with glycolysis. These 4 define what is unique about gluconeogenesis. Any question asking “which enzyme is found in gluconeogenesis but NOT glycolysis?” — one of these 4 is the answer.
🔴 PEPCK is the most important regulated and transcriptionally controlled enzyme of gluconeogenesis Induced by glucagon (cAMP) and cortisol; suppressed by insulin (FOXO1 pathway). It is the primary target of drugs and hormones that control blood glucose.
🔴 Pyruvate carboxylase requires Biotin and is activated by Acetyl-CoA When Acetyl-CoA is high (active fat oxidation in fasting), pyruvate carboxylase is signalled to convert pyruvate to OAA for gluconeogenesis. This is the key link between fat oxidation and gluconeogenesis activation.
🔴 Glucose-6-phosphatase is present only in liver and kidney — NOT in muscle or brain This is why only liver and kidney can release free glucose into the blood. Muscle cannot complete gluconeogenesis to free glucose.
🔴 Fructose-2,6-bisphosphate is the most potent allosteric regulator of both glycolysis AND gluconeogenesis High F-2,6-BP → glycolysis ON (activates PFK-1), gluconeogenesis OFF (inhibits FBPase-1). Low F-2,6-BP → reverse. Glucagon lowers it; insulin raises it.
🔴 Even-chain fatty acids CANNOT contribute to net gluconeogenesis; odd-chain fatty acids CAN (via propionyl-CoA → succinyl-CoA) The mechanism: Acetyl-CoA carbons are lost as CO₂ in TCA; there is no net new OAA. Propionyl-CoA → succinyl-CoA IS a net anaplerotic reaction.
🟠 The energy cost of gluconeogenesis from 2 pyruvate → 1 glucose = 4 ATP + 2 GTP = 6 ATP equivalents This energy comes from fatty acid oxidation during fasting. Gluconeogenesis is expensive — the body runs it only when essential.
🟠 Von Gierke disease (GSD Type Ia) = G6Pase deficiency → blocks both glycogenolysis AND gluconeogenesis Clinical triad: severe fasting hypoglycaemia + hepatomegaly + lactic acidosis. Plus hyperlipidaemia and hyperuricaemia.
🟠 The Cori cycle transfers the energy burden of anaerobic muscle metabolism to the liver Muscle produces lactate → liver converts it to glucose (at cost of 6 ATP from fat oxidation) → muscle uses glucose again. The liver subsidises anaerobic muscle metabolism.
🟠 Glucagon phosphorylates PFK-2/FBPase-2 → activates FBPase-2 domain → lowers F-2,6-BP → gluconeogenesis ON This is the complete hormonal signalling chain for fasting activation of gluconeogenesis. Follow each step in sequence.
🟠 Metformin lowers blood glucose by inhibiting hepatic gluconeogenesis (via Complex I inhibition → AMPK activation) Not by stimulating insulin secretion. Metformin is not an insulin secretagogue — this is frequently confused with sulfonylureas.
🟡 Purely ketogenic amino acids = Leucine + Lysine ONLY — they cannot contribute to gluconeogenesis All other amino acids are at least partly glucogenic. “Leu and Lys alone = Ketogenic alone” is the rule. Phenylalanine, isoleucine, tyrosine, tryptophan, threonine are BOTH glucogenic AND ketogenic.
🟡 Alcohol causes hypoglycaemia by depleting pyruvate and OAA via NADH excess High NADH (from ethanol oxidation) drives pyruvate → lactate and OAA → malate, removing both key gluconeogenic substrates. Hypoglycaemia in fasting alcoholics is a direct biochemical consequence.
🟡 Cortisol is the most important inducer of PEPCK transcription In Cushing syndrome or steroid therapy, PEPCK is chronically upregulated → persistent gluconeogenesis → fasting hyperglycaemia → steroid-induced diabetes.
🧠 Mnemonics & Memory Tricks
“People Can Go Places” — The 4 Unique Gluconeogenic Enzymes → Helps you remember all 4 unique enzymes in order of the pathway
P = Pyruvate carboxylase (Pyruvate → OAA) C = CPEPCK / Carboxykinase (OAA → PEP) G = FBPase-1 — Gluco… neogenesis bypass (F-1,6-BP → F-6-P) P = Glucose-6–Phosphatase (G6P → Glucose)
Alternative version that is cleaner:
“Private Cars Go Places” P = Pyruvate carboxylase C = CPEPCK (PEP Carboxykinase) G = FBPase-1 (Fructose bisphosphatase — the Gate before glucose) P = Glucose-6-Phosphatase
💡 Pro tip: In any exam question asking “which of the following is unique to gluconeogenesis?” — one of these 4 enzymes is correct. If you see pyruvate carboxylase, PEPCK, FBPase-1, or glucose-6-phosphatase — that is a gluconeogenesis-specific enzyme.
“LOLA Likes Keto” — The Purely Ketogenic Amino Acids → Helps you remember that only Leucine and Lysine are purely ketogenic (cannot contribute to gluconeogenesis)
L = Leucine O = Only these two L = Lysine A = Are purely ketogenic
💡 Pro tip: Every other amino acid has at least some glucogenic potential. When a question asks “which amino acid(s) CANNOT contribute to gluconeogenesis?” — the answer is Leucine and Lysine.
“Glucagon Lowers F-2,6-BP; Insulin Raises It — Opposite Effects, Opposite States” → Helps you remember the hormonal control of the master regulator
Glucagon → PKA → phosphorylates PFK-2/FBPase-2 → FBPase-2 active → F-2,6-BP falls → Gluconeogenesis ON Insulin → phosphatase → dephosphorylates PFK-2/FBPase-2 → PFK-2 active → F-2,6-BP rises → Glycolysis ON
Memory hook: “Glucagon Grounds the F-2,6-BP plane” — glucagon brings F-2,6-BP down to the ground (low level).
💡 Pro tip: F-2,6-BP is the single most testable regulatory molecule linking glycolysis and gluconeogenesis. Draw the bifunctional enzyme diagram once from memory. Every exam has at least one question testing this axis.
“Biotin Carries CO₂ — All Carboxylases Need Biotin” → Pyruvate carboxylase, Acetyl-CoA carboxylase (FA synthesis), Propionyl-CoA carboxylase, Methylcrotonyl-CoA carboxylase — ALL require biotin
💡 Pro tip: Any question involving “CO₂ fixation” or “carboxylation” in metabolic pathways → the answer involves biotin. Biotin deficiency or biotinidase deficiency → failure of ALL carboxylase reactions → multiple simultaneous pathway failures.
⚠️ Common Mistakes Students Make on This Topic
❌ Mistake: “Gluconeogenesis is just glycolysis running in reverse” ✅ Reality: 7 of 10 steps are shared (and are reversible), but 3 thermodynamic barriers require 4 completely different bypass enzymes. Without pyruvate carboxylase, PEPCK, FBPase-1, and glucose-6-phosphatase, gluconeogenesis cannot occur. 📝 How this gets tested: “Which of the following enzymes participates in BOTH glycolysis and gluconeogenesis?” — the shared reversible enzymes (e.g., enolase, aldolase, phosphoglycerate kinase) are the answer. Pyruvate kinase and hexokinase are NOT shared.
❌ Mistake: “Fatty acids are good substrates for gluconeogenesis” ✅ Reality: Even-chain fatty acids (the vast majority) cannot contribute to net gluconeogenesis because Acetyl-CoA carbons are lost as CO₂ in the TCA cycle. Only the glycerol backbone of fat and propionyl-CoA from odd-chain fatty acids contribute. 📝 How this gets tested: “A patient is fasting — which of the following can contribute to de novo glucose synthesis?” — fatty acids listed as an option is a TRAP. Glycerol is correct; fatty acids (even-chain) are not.
❌ Mistake: “Metformin stimulates insulin secretion” ✅ Reality: Metformin does NOT stimulate insulin secretion. It primarily inhibits hepatic gluconeogenesis (via Complex I → AMPK pathway). It does not cause hypoglycaemia when used as monotherapy because it does not raise insulin. 📝 How this gets tested: “Which anti-diabetic drug works by reducing hepatic glucose production without stimulating insulin secretion?” — metformin. “Which drug can cause hypoglycaemia as monotherapy?” — NOT metformin (sulfonylureas do).
❌ Mistake: “The Cori cycle generates net energy for the body” ✅ Reality: The Cori cycle is a net energy-consuming process at the whole-body level. The liver expends 6 ATP to reconvert 2 lactate to 1 glucose, while muscle only gained 2 ATP when it converted that glucose to lactate. Net loss = 4 ATP per cycle. The energy comes from fat oxidation in the liver. 📝 How this gets tested: “What is the net energetic consequence of the Cori cycle?” — the answer is net ATP consumption (subsidised by hepatic fat oxidation), not ATP generation.
❌ Mistake: “Pyruvate carboxylase converts pyruvate directly to PEP” ✅ Reality: Pyruvate carboxylase converts pyruvate → OAA only. It is PEPCK that converts OAA → PEP. These are two separate enzymes, two separate steps, requiring two separate energy equivalents (ATP for pyruvate carboxylase; GTP for PEPCK). 📝 How this gets tested: Enzyme-matching questions where “pyruvate carboxylase” and “PEPCK” are listed separately. Confusing them or attributing the wrong reaction to each is a common exam error.
📝 5 Practice MCQs — Test Yourself Now
Q1: Which of the following enzymes is present in the liver but ABSENT in skeletal muscle, explaining why muscle cannot release free glucose into the bloodstream?
- A. Phosphoglycerate kinase
- B. Glucose-6-phosphatase
- C. Fructose-1,6-bisphosphatase
- D. Pyruvate carboxylase
✅ Answer: B. Glucose-6-phosphatase
Why correct: Glucose-6-phosphatase (G6Pase) is expressed only in liver and kidney cortex, not in skeletal muscle (or brain). It catalyses the final step: Glucose-6-phosphate → Glucose + Pi. Without G6Pase, glucose-6-phosphate cannot be converted to free glucose and remains trapped inside the cell. Muscle can phosphorylate glucose but cannot dephosphorylate it back — it uses glucose only for its own energy, never for export.
Why A is wrong: Phosphoglycerate kinase is a shared enzyme of both glycolysis and gluconeogenesis, present in virtually all tissues. It has no role in glucose export. Why C is wrong: FBPase-1 is expressed in liver and also in muscle to some extent, but even if muscle had FBPase-1, it still cannot release glucose without G6Pase. Why D is wrong: Pyruvate carboxylase is present in multiple tissues including muscle (though predominantly liver and kidney). Its absence in muscle does not explain the inability to export glucose.
Exam tip: “Glucose release into blood” = G6Pase = liver + kidney only. Von Gierke disease (G6Pase deficiency) blocks both glycogenolysis AND gluconeogenesis from releasing glucose — the classic example of why this enzyme is essential.
Q2: A 14-month-old child presents with severe fasting hypoglycaemia, massive hepatomegaly, lactic acidosis, and elevated triglycerides. Liver biopsy shows massive glycogen accumulation. Which enzyme deficiency is most likely?
- A. Glycogen phosphorylase (liver)
- B. Glucose-6-phosphatase
- C. Fructose-1,6-bisphosphatase
- D. Branching enzyme
✅ Answer: B. Glucose-6-phosphatase
Why correct: This is the classic presentation of Von Gierke disease (GSD Type Ia). G6Pase deficiency means that glucose-6-phosphate — produced by both glycogenolysis AND gluconeogenesis — cannot be converted to free glucose. G6P accumulates, driving: glycogen synthesis (hepatomegaly), glycolysis → pyruvate → lactate (lactic acidosis), de novo lipogenesis (hypertriglyceridaemia), PPP → NADPH excess. Severe fasting hypoglycaemia results because neither glycogenolysis NOR gluconeogenesis can rescue blood glucose.
Why A is wrong: Glycogen phosphorylase (liver) deficiency = GSD Type VI (Hers disease) — much milder, causes hepatomegaly but only moderate hypoglycaemia. Lactate is NOT elevated because gluconeogenesis is intact. Why C is wrong: FBPase-1 deficiency also causes fasting hypoglycaemia and lactic acidosis but does NOT cause massive glycogen accumulation (hepatomegaly from glycogen is not a feature — liver size is less pronounced). Why D is wrong: Branching enzyme deficiency = GSD Type IV (Andersen disease) — causes liver disease (cirrhosis, liver failure) but NOT the specific metabolic pattern of hypoglycaemia + lactic acidosis + hypertriglyceridaemia.
Exam tip: Von Gierke = G6Pase deficiency = GSD Type Ia = the only GSD that blocks BOTH glycogenolysis AND gluconeogenesis simultaneously. The tetrad of fasting hypoglycaemia + hepatomegaly + lactic acidosis + hypertriglyceridaemia is pathognomonic.
Q3: During prolonged fasting, fatty acid oxidation in the liver increases markedly. Which of the following best explains how this directly stimulates gluconeogenesis?
- A. NADH produced by beta-oxidation activates pyruvate carboxylase by reducing its NAD⁺ requirement
- B. Acetyl-CoA produced by beta-oxidation allosterically activates pyruvate carboxylase AND inhibits PDH, redirecting pyruvate toward gluconeogenesis
- C. Increased ATP from beta-oxidation activates PFK-1, accelerating glycolysis and providing gluconeogenic substrate
- D. Acetyl-CoA directly enters the gluconeogenic pathway as a 2-carbon gluconeogenic unit
✅ Answer: B. Acetyl-CoA produced by beta-oxidation allosterically activates pyruvate carboxylase AND inhibits PDH, redirecting pyruvate toward gluconeogenesis
Why correct: This is the elegant molecular linkage between fat oxidation and gluconeogenesis. Rising Acetyl-CoA from beta-oxidation does two things simultaneously: (1) Allosterically activates pyruvate carboxylase → pyruvate → OAA → feeds gluconeogenesis; (2) Inhibits PDH → prevents pyruvate from entering the TCA cycle directly. The net effect is that pyruvate is redirected away from the TCA cycle and toward gluconeogenesis. ATP from fat oxidation then provides the energy for the expensive steps (pyruvate carboxylase and PEPCK).
Why A is wrong: NADH does not activate pyruvate carboxylase. The activation is by Acetyl-CoA, not NADH. High NADH actually impairs gluconeogenesis by depleting pyruvate (driving pyruvate → lactate) and OAA (driving OAA → malate). Why C is wrong: ATP activates FBPase-1 (gluconeogenesis) and INHIBITS PFK-1 (glycolysis) — not the other way around. Activating glycolysis would consume glucose, not produce it. Why D is wrong: This is the classic misconception. Acetyl-CoA CANNOT be a net gluconeogenic substrate in mammals. Its carbons are lost as CO₂ in the TCA cycle. It activates gluconeogenesis (via pyruvate carboxylase) but does not contribute its own carbons to glucose.
Exam tip: “Why does fasting fat oxidation stimulate gluconeogenesis?” → Acetyl-CoA activates pyruvate carboxylase (allosteric) AND inhibits PDH (product inhibition). Both effects push pyruvate toward OAA and gluconeogenesis.
Q4: A 52-year-old man with type 2 diabetes and stage 3 chronic kidney disease (eGFR 28 mL/min) is brought in with altered consciousness and blood pH of 7.18. Lactate is 8 mmol/L (normal <2). He has been taking metformin for 5 years. What is the primary biochemical mechanism of his condition?
- A. Metformin stimulates insulin secretion, causing hypoglycaemia-induced lactic acidosis
- B. Metformin inhibits Complex I of the ETC → impairs NADH reoxidation → shifts pyruvate → lactate; renal failure prevents lactate clearance
- C. Metformin inhibits glucokinase, causing glucose-6-phosphate accumulation and lactic acidosis
- D. Renal failure reduces insulin clearance, causing insulin excess and increased lactate production
✅ Answer: B. Metformin inhibits Complex I of the ETC → impairs NADH reoxidation → shifts pyruvate → lactate; renal failure prevents lactate clearance
Why correct: Metformin’s primary mechanism is inhibition of Complex I (NADH:ubiquinone oxidoreductase) of the mitochondrial ETC in hepatocytes. This impairs NADH reoxidation → cytoplasmic NADH/NAD⁺ ratio rises → LDH is driven toward converting pyruvate to lactate. Simultaneously, less ATP is produced → pyruvate carboxylase and PEPCK are starved of energy → gluconeogenesis falls (the therapeutic effect). In normal circumstances, the liver clears lactate continuously via gluconeogenesis. With renal failure, metformin accumulates (it is renally excreted) → Complex I inhibition is amplified → AND kidneys cannot clear lactate independently → lactic acidosis develops.
Why A is wrong: Metformin does NOT stimulate insulin secretion — this is a defining characteristic that distinguishes it from sulfonylureas. Metformin monotherapy does not cause hypoglycaemia. Why C is wrong: Metformin has no direct effect on glucokinase. Its target is Complex I of the ETC (and AMPK activation secondarily). Why D is wrong: Renal failure does reduce insulin clearance, potentially causing hypoglycaemia, but this is not the mechanism of metformin-associated lactic acidosis, which is specifically due to Complex I inhibition and lactate accumulation.
Exam tip: Metformin + renal impairment (eGFR <30) = contraindication because of lactic acidosis risk. The mechanism: Complex I inhibition → ↑NADH/NAD⁺ → pyruvate → lactate, compounded by reduced renal drug excretion and impaired renal lactate clearance.
Q5: In the glucose-alanine cycle, which of the following correctly describes the biochemical events occurring in the LIVER?
- A. Alanine is deaminated to ammonia and pyruvate; ammonia enters the urea cycle; pyruvate is used for gluconeogenesis
- B. Alanine is transaminated with α-ketoglutarate → pyruvate + glutamate; glutamate enters urea cycle; pyruvate → gluconeogenesis
- C. Alanine is directly phosphorylated to glucose-6-phosphate by hexokinase without intermediate steps
- D. Alanine is converted to acetyl-CoA and oxidised in the TCA cycle to provide energy for gluconeogenesis
✅ Answer: B. Alanine is transaminated with α-ketoglutarate → pyruvate + glutamate; glutamate enters urea cycle; pyruvate → gluconeogenesis
Why correct: In the liver, alanine is processed by the enzyme alanine aminotransferase (ALT/GPT): Alanine + α-Ketoglutarate → Pyruvate + Glutamate. The glutamate then enters the urea cycle (via glutamate dehydrogenase releasing NH₄⁺, or via transamination to aspartate which enters the urea cycle directly). The pyruvate enters gluconeogenesis (pyruvate carboxylase → OAA → PEPCK → PEP → … → Glucose). The glucose is exported back to muscle. This cycle simultaneously handles nitrogen disposal (urea cycle) and glucose production (gluconeogenesis) in the liver.
Why A is wrong: Alanine is NOT deaminated (which would directly release ammonia); it is transaminated — the amino group is transferred to α-ketoglutarate, not released as free NH₃ in the first step. This distinction is important because free ammonia is toxic and must be handled carefully. Why C is wrong: Alanine cannot be directly phosphorylated to glucose-6-phosphate. It must first be converted to pyruvate, then go through gluconeogenesis step by step. Why D is wrong: Alanine is NOT converted to acetyl-CoA. Pyruvate → Acetyl-CoA (via PDH) would be the ketogenic/oxidative fate. In gluconeogenesis, pyruvate carboxylase converts pyruvate to OAA, explicitly bypassing PDH.
Exam tip: ALT (alanine aminotransferase) = the enzyme of the glucose-alanine cycle in the liver. Elevated ALT in liver disease reflects disruption of this and other transamination reactions. The reaction is: Alanine + α-KG → Pyruvate + Glutamate (catalysed by ALT, requires pyridoxal phosphate/B6).
📚 References
📖 Harper’s Illustrated Biochemistry — Rodwell et al. | Chapter 19: Gluconeogenesis & the Control of Blood Glucose
📖 Lippincott’s Illustrated Reviews: Biochemistry — Ferrier | Chapter 10: Gluconeogenesis
📖 Lehninger Principles of Biochemistry — Nelson & Cox | Chapter 15: Glucose Catabolism; Chapter 23: Biosynthesis of Amino Acids, Nucleotides & Related Molecules (for glucogenic amino acids)
📖 Stryer’s Biochemistry — Berg, Tymoczko & Stryer | Chapter 16: Glycolysis and Gluconeogenesis (unified chapter)
📖 Vasudevan & Sreekumari’s Textbook of Biochemistry — Vasudevan | Chapter 8: Metabolism of Carbohydrates — Gluconeogenesis
🚀 Keep Practising — You Are Not Done Yet
Gluconeogenesis questions in NEET PG and USMLE rarely come as isolated pathway questions. They come as clinical scenarios — a fasting child with hypoglycaemia, an alcoholic with lactic acidosis, a diabetic on metformin with renal failure. The biochemistry is the tool for understanding the clinical picture.
The only way to develop that clinical-biochemistry integration is through structured practice under timed conditions.
medicalmcq.in has free Biochemistry MCQs covering gluconeogenesis, glycolysis, the Cori cycle, and every pathway that connects to this topic — all with detailed mechanistic explanations, not just answer keys.
After reading this article, do at least 15–20 gluconeogenesis MCQs immediately. Then revisit in 48 hours. That is how biochemistry moves from reading to examination performance.