What You Will Learn in This Article
- Name all 9 essential amino acids and recall the mnemonic to never forget them
- Explain the key metabolic roles of each essential amino acid with clinical relevance
- Classify every amino acid as glucogenic, ketogenic, or both — and understand the metabolic logic
- Trace the major degradation pathways: transamination, deamination, and entry into TCA cycle intermediates
- Connect essential amino acids to neurotransmitter synthesis, haem production, one-carbon metabolism, and hormone synthesis
- Identify the specific diseases caused by defects in essential amino acid metabolism (PKU, MSUD, homocystinuria, Hartnup disease)
- Answer clinical-scenario MCQs on amino acid metabolism confidently
📖 Introduction: Why This Topic Matters in Exams
A 3-month-old infant is brought to the clinic with progressive intellectual disability, a musty odour to the urine, and fair skin despite dark-haired parents. The paediatrician suspects a metabolic disorder. Blood phenylalanine is markedly elevated. The diagnosis is phenylketonuria — a disorder of phenylalanine metabolism caused by deficiency of phenylalanine hydroxylase, the enzyme that converts the essential amino acid phenylalanine to tyrosine. Without early dietary intervention — a low-phenylalanine diet — the accumulated phenylalanine is neurotoxic, and the child suffers irreversible cognitive damage. This is not an abstract biochemistry problem. It is a disease where understanding amino acid metabolism directly saves children’s lives.
Amino acid metabolism is tested relentlessly in NEET PG and USMLE Step 1. Questions come in pure recall form (“which amino acids are essential?”), mechanistic form (“why does MSUD cause lactic acidosis?”), and full clinical scenarios (“a child with recurrent encephalopathy and sweet-smelling urine”). The clinical diseases linked to essential amino acid metabolism — PKU, MSUD, homocystinuria, Hartnup disease, albinism, tyrosinaemia — are among the most high-yield inborn errors of metabolism in any exam.
This article covers all 9 essential amino acids systematically — their structure, metabolic roles, degradation pathways, and the diseases that result when each goes wrong. By the end, you will have the clinical and biochemical framework to handle any exam question on this topic.
🔬 Section 1 — Classification of Amino Acids: Essential vs Non-Essential
The Essential Amino Acids — Why “Essential”?
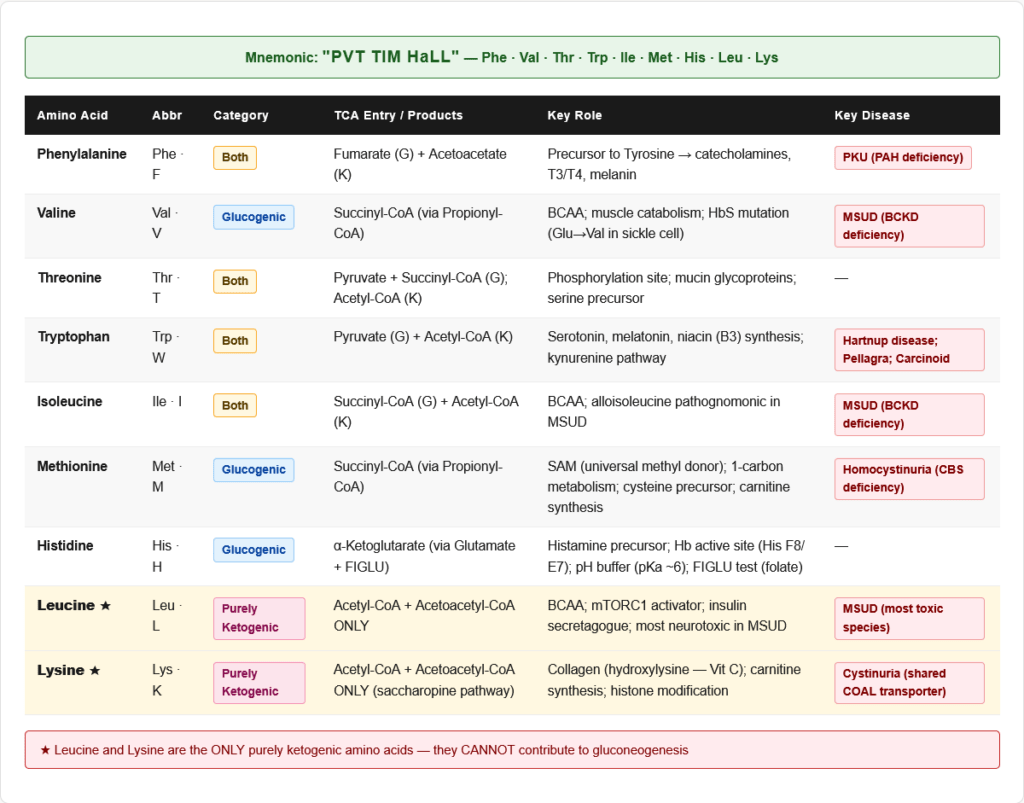
An amino acid is essential (indispensable) if the body cannot synthesise it in sufficient quantities to meet physiological needs and it must therefore be obtained from the diet. The designation is not permanent — it reflects the metabolic capacity of the human body under normal conditions.
The 9 Essential Amino Acids:
| Amino Acid | Abbreviation | Single-letter code |
|---|---|---|
| Histidine | His | H |
| Isoleucine | Ile | I |
| Leucine | Leu | L |
| Lysine | Lys | K |
| Methionine | Met | M |
| Phenylalanine | Phe | F |
| Threonine | Thr | T |
| Tryptophan | Trp | W |
| Valine | Val | V |
Mnemonic: “PVT TIM HaLL” P = Phenylalanine, V = Valine, T = Threonine, T = Tryptophan, I = Isoleucine, M = Methionine, H = Histidine, L = Leucine, L = Lysine
Conditionally Essential Amino Acids
Some amino acids become essential under conditions of illness, prematurity, or increased demand:
| Amino Acid | Condition Making It Essential |
|---|---|
| Arginine | Rapid growth, surgery, sepsis, urea cycle disorders |
| Cysteine | Premature neonates (cannot synthesise from methionine); liver disease |
| Tyrosine | PKU (phenylalanine hydroxylase deficiency — cannot make from Phe) |
| Glutamine | Critical illness, trauma, cancer cachexia |
| Proline | Severe illness, wound healing |
| Serine | Certain neurological conditions |
| Glycine | Premature neonates |
Glucogenic, Ketogenic, and Mixed Classification
This classification tells you what TCA/gluconeogenic intermediates an amino acid produces on catabolism:
Purely Ketogenic (2 only — MUST MEMORISE):
- Leucine — produces Acetyl-CoA + Acetoacetyl-CoA only
- Lysine — produces Acetyl-CoA + Acetoacetyl-CoA only
Both Glucogenic AND Ketogenic (5 — “PIT TF”):
- Phenylalanine → Fumarate (glucogenic) + Acetoacetate (ketogenic)
- Isoleucine → Succinyl-CoA (glucogenic) + Acetyl-CoA (ketogenic)
- Tryptophan → Pyruvate/Alanine (glucogenic) + Acetyl-CoA (ketogenic)
- Tyrosine → Fumarate (glucogenic) + Acetoacetate (ketogenic)
- Threonine → Pyruvate/Succinyl-CoA (glucogenic) + Acetyl-CoA (ketogenic)
Purely Glucogenic (all remaining):
- Valine → Succinyl-CoA
- Methionine → Succinyl-CoA
- Histidine → α-Ketoglutarate (via Glutamate)
🔬 Section 2 — Each Essential Amino Acid: Structure, Roles, and Metabolism
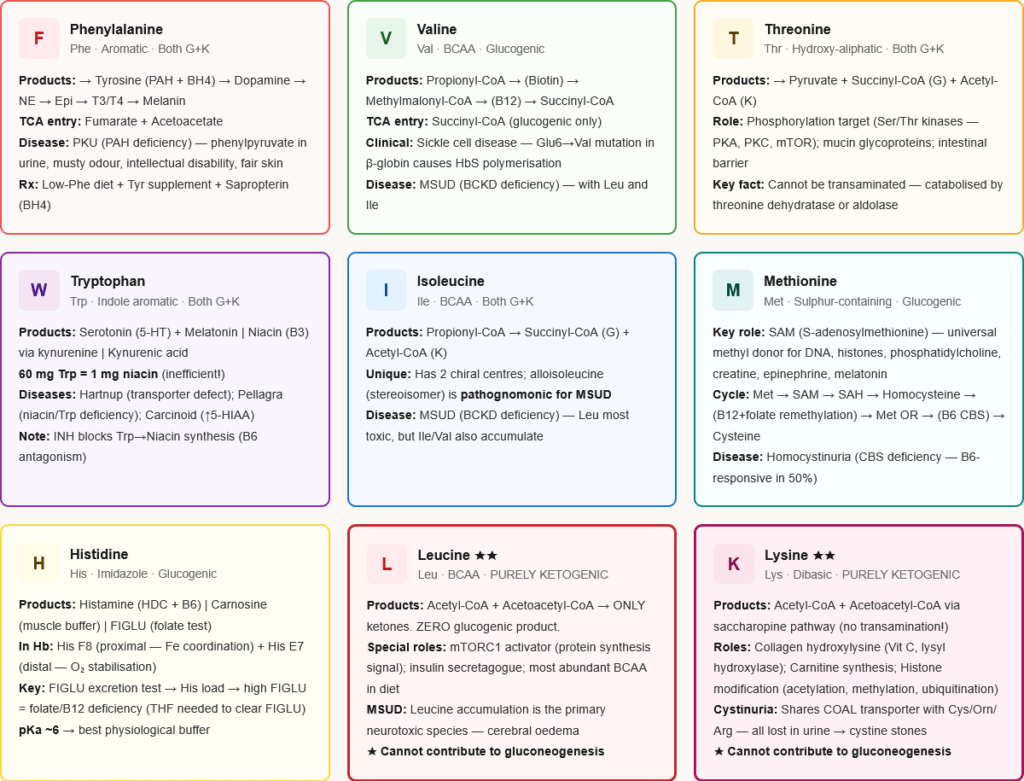
1. Phenylalanine (Phe, F)
Structure: Aromatic — benzyl group on alanine backbone
Key metabolic roles:
- Precursor to tyrosine (via phenylalanine hydroxylase — requires tetrahydrobiopterin/BH4 as cofactor)
- Tyrosine then produces: DOPA → Dopamine → Norepinephrine → Epinephrine; Thyroid hormones (T3/T4); Melanin
- Carbon skeleton enters as fumarate (glucogenic) and acetoacetate (ketogenic)
Rate-limiting enzyme of catabolism: Phenylalanine hydroxylase (PAH)
Key disease:
- Phenylketonuria (PKU): PAH deficiency → phenylalanine accumulates → converted to phenylpyruvate (phenylketone) → excreted in urine → musty/mousy odour. Neurotoxic: inhibits aromatic amino acid transport across BBB → depletes neurotransmitters. Treatment: low-Phe diet + tyrosine supplementation (tyrosine becomes essential). Sapropterin (BH4 analogue) for BH4-responsive PKU.
- Malignant hyperphenylalaninaemia: BH4 deficiency (dihydropteridine reductase or pterin synthesis defects) — PAH is intact but cannot function without BH4. Affects all BH4-dependent enzymes: PAH, tyrosine hydroxylase, tryptophan hydroxylase → depletes all catecholamines AND serotonin. More severe than classic PKU.
2. Tyrosine (Tyr, Y) — Conditionally Essential
Structure: Aromatic — hydroxyphenyl group (hydroxylated phenylalanine)
Note: Non-essential under normal conditions (synthesised from phenylalanine). Becomes essential in PKU.
Key metabolic roles — The most productive amino acid in the body:
| Product | Pathway | Clinical relevance |
|---|---|---|
| DOPA → Dopamine | Tyrosine hydroxylase (rate-limiting; BH4 required) | Parkinson disease (dopamine depletion) |
| Dopamine → Norepinephrine | Dopamine β-hydroxylase | Pheochromocytoma; stress response |
| Norepinephrine → Epinephrine | PNMT (in adrenal medulla; cortisol-induced) | Flight-or-fight response |
| Thyroid hormones (T3, T4) | Thyroid peroxidase (iodination of tyrosine) | Hypothyroidism; Graves disease |
| Melanin | Tyrosinase (Cu-dependent) | Albinism (tyrosinase deficiency) |
| Fumarate + Acetoacetate | Fumarylacetoacetase | Tyrosinaemia Type I (HT-1) |
Key diseases:
- Tyrosinaemia Type I (HT-1): Fumarylacetoacetase deficiency → fumarylacetoacetate + succinylacetone accumulate → liver/kidney damage, cirrhosis, hepatocellular carcinoma risk. Succinylacetone inhibits ALA dehydratase → porphyria-like crises. Treatment: NTBC (nitisinone) + low-phenylalanine/tyrosine diet
- Albinism: Tyrosinase deficiency → cannot produce melanin → hypopigmentation of skin/hair/eyes, photophobia, nystagmus, increased skin cancer risk
- Alkaptonuria: Homogentisate oxidase deficiency → homogentisic acid accumulates → excreted in urine (turns black on standing — ochronuria) → deposits in cartilage/tendons (ochronosis) → arthritis. Urine darkens on exposure to air/alkali.
3. Tryptophan (Trp, W)
Structure: Aromatic — indole ring system; largest of the essential amino acids
Key metabolic roles — The most versatile:
| Product | Enzyme/Pathway | Notes |
|---|---|---|
| Serotonin (5-HT) | Tryptophan hydroxylase (rate-limiting; BH4) → AADC (B6) | Rate-limited by tryptophan availability; mood regulation; gut motility |
| Melatonin | From serotonin → N-acetyltransferase → HIOMT | Circadian rhythm; pineal gland |
| Niacin (Vitamin B3) | Kynurenine pathway → quinolinate → NAD⁺ | 60 mg Trp = 1 mg niacin (inefficient). Explains why corn-based diets (low Trp) cause pellagra even if total protein adequate. |
| Kynurenic acid | Kynurenine aminotransferase | NMDA receptor antagonist; increased in schizophrenia |
| Tryptamine / indole acetic acid | Gut bacteria metabolism |
Key diseases:
- Pellagra (Niacin deficiency): Caused by niacin-deficient diet OR tryptophan deficiency (corn/maize diets) OR isoniazid (INH — competes with pyridoxal phosphate, blocking niacin synthesis from Trp). Classic 4 Ds: Dermatitis, Diarrhoea, Dementia, Death. Dermatitis is photosensitive (Casal’s necklace — necklace-like distribution on neck).
- Hartnup Disease: Autosomal recessive defect in neutral amino acid transporter (SLC6A19) in intestine AND kidney. Tryptophan (and other neutral amino acids) cannot be absorbed from gut. Pellagra-like presentation (because Trp → niacin pathway is disrupted), cerebellar ataxia, photosensitive rash. Treated with nicotinamide supplementation.
- Carcinoid Syndrome: Neuroendocrine tumour (especially ileal) produces excess serotonin from tryptophan → flushing, diarrhoea, bronchoconstriction, right-sided heart lesions (tricuspid/pulmonary). Diagnosis: urinary 5-HIAA (5-hydroxyindoleacetic acid — serotonin metabolite). Pellagra can occur because so much Trp is diverted to serotonin that niacin synthesis is depleted.
4. Methionine (Met, M)
Structure: Sulphur-containing; S-methyl side chain
Key metabolic roles — The master methyl donor:
Methionine’s central metabolic role is the one-carbon (methyl group) transfer cycle:
The Methionine Cycle:
- Methionine + ATP → S-Adenosylmethionine (SAM) — enzyme: Methionine adenosyltransferase
- SAM donates its methyl group to a substrate → S-Adenosylhomocysteine (SAH)
- SAH → Homocysteine + Adenosine — enzyme: SAH hydrolase
- Homocysteine has two fates:
- Remethylation to Methionine: Homocysteine + 5-methylTHF → Methionine — enzyme: Methionine synthase (requires Vitamin B12 as cofactor and folate as methyl donor)
- Transsulphuration to Cysteine: Homocysteine + Serine → Cystathionine (enzyme: cystathionine β-synthase, requires B6) → Cysteine
SAM methylation targets:
- DNA methylation (gene regulation, epigenetics)
- Histone methylation
- Phosphatidylcholine synthesis (from phosphatidylethanolamine)
- Creatine synthesis (Arg + Gly → guanidinoacetate, then SAM methylates → Creatine)
- Norepinephrine → Epinephrine (PNMT uses SAM)
- Serotonin → Melatonin (N-acetyltransferase + HIOMT, SAM involved)
- mRNA capping (5′ cap)
Key diseases:
- Homocystinuria (Classical): Cystathionine β-synthase (CBS) deficiency → Homocysteine accumulates → cannot enter transsulphuration → elevated plasma homocysteine. Presentation: Marfanoid habitus (tall, long limbs), ectopia lentis (downward displacement — unlike Marfan which is upward), intellectual disability, thromboembolic events (Hcy is prothrombotic — damages endothelium, activates coagulation). Treatment: B6 supplementation (if B6-responsive CBS mutation), low-methionine diet, folate + B12 (to push remethylation), betaine (alternative methyl donor for remethylation).
- Hyperhomocysteinaemia (Mild-Moderate): B12 or folate deficiency → methionine synthase fails → homocysteine rises. Cardiovascular risk factor. Common in elderly with poor nutrition.
- MTHFR Deficiency: Methylenetetrahydrofolate reductase (MTHFR) deficiency → cannot make 5-methyl THF → methionine synthase lacks substrate → homocysteine rises even if B12 normal. Associated with neural tube defects (folate-sensitive).
5. Histidine (His, H)
Structure: Imidazole side chain (pKa ~6.0 — acts as buffer at physiological pH)
Key metabolic roles:
- Precursor to histamine (via histidine decarboxylase — requires pyridoxal phosphate/B6)
- Histamine: mediates allergic responses (H1), gastric acid secretion (H2), neurotransmission (H3/H4)
- Precursor to carnosine (β-alanyl-histidine) — dipeptide buffer in muscle; antioxidant
- Component of haemoglobin — the proximal and distal histidines (His F8 and His E7) are critical for oxygen binding; His F8 (proximal) coordinates the haem iron; His E7 (distal) stabilises O₂ binding
- Part of the active site of many enzymes — the imidazole ring is the most versatile catalytic group: can act as acid, base, or nucleophile at physiological pH
- Catabolism: Histidine → urocanate → N-formiminoglutamate (FIGLU) → glutamate → α-ketoglutarate (TCA). The FIGLU step requires folate (as tetrahydrofolate acceptor)
FIGLU excretion test:
- Histidine load → measure urinary FIGLU
- High FIGLU after histidine load = folate deficiency (folate needed to convert FIGLU → glutamate)
- Also elevated in B12 deficiency (because B12 is needed to regenerate active folate)
Key fact: Histidine is essential in infants and growing children (growth demands exceed synthetic capacity) but debated in adults. Most texts and all major exams list it as essential.
6. Lysine (Lys, K)
Structure: Diaminomonocarboxylic acid — two amino groups; strongly basic
Key metabolic roles:
- Collagen: Lysine hydroxylation (lysyl hydroxylase — requires Vitamin C and iron) → hydroxylysine → cross-linking of collagen fibrils → tensile strength. Vitamin C deficiency (Scurvy) impairs lysyl hydroxylase → fragile collagen → bleeding gums, perifollicular haemorrhages, wound dehiscence, poor wound healing
- Carnitine synthesis: Lysine + methionine → carnitine (via a 5-step pathway requiring Vitamin C, niacin, B6, and iron). Carnitine is essential for long-chain fatty acid transport into mitochondria. Deficiency → cardiomyopathy, hypoketotic hypoglycaemia.
- Histone modification: Lysine residues on histones are acetylated, methylated, or ubiquitinated — critical for gene regulation
- Crosslinking proteins: Fibrin crosslinking (Factor XIIIa), elastin crosslinking (lysyl oxidase — Cu-dependent)
- Catabolism: Unique — lysine is catabolised via the saccharopine pathway (not by transamination — it has no α-keto acid equivalent via transamination) → eventually produces Acetyl-CoA + Acetoacetyl-CoA → purely ketogenic
Key fact: Lysine and Arginine share the same intestinal and renal transporter (CAT-1/SLC7A1). Excess arginine competitively inhibits lysine absorption — relevant in lysine supplementation and in cystinuria (where the shared transporter for Cys, Orn, Arg, Lys is defective).
Key disease:
- Cystinuria: Defect in shared renal/intestinal transporter for Cystine, Ornithine, Arginine, Lysine (COAL mnemonic) → these amino acids are lost in urine → cystine stones (poorly soluble) → renal calculi. Note: plasma levels are NORMAL (only renal wasting). Treatment: high fluid intake, alkalinise urine (cystine soluble at pH >7.5), D-penicillamine, tiopronin.
7. Leucine (Leu, L)
Structure: Branched-chain aliphatic amino acid (BCAA)
Key metabolic roles:
- One of the 3 BCAAs (with Isoleucine and Valine) — uniquely catabolised in muscle (not liver) via BCAA aminotransferase
- mTORC1 activator — leucine is the most potent dietary activator of mTOR signalling → protein synthesis, cell growth. This is the scientific basis for leucine-rich protein supplements in muscle building and sarcopenia management.
- Insulin secretagogue — directly stimulates β-cell insulin release (along with glucose)
- Ketogenesis: Leucine is the most ketogenic amino acid — produces Acetyl-CoA and Acetoacetyl-CoA entirely; no glucogenic product. In MSUD, excess leucine is the primary neurotoxic species.
- Catabolism: Leucine → Isovaleryl-CoA → HMG-CoA → Acetoacetate + Acetyl-CoA (entirely ketogenic)
Key disease:
- Maple Syrup Urine Disease (MSUD): Deficiency of branched-chain α-ketoacid dehydrogenase (BCKD) complex → all 3 BCAAs (Leu, Ile, Val) and their α-keto acids accumulate → urine smells of maple syrup/caramel. The BCKD complex uses same cofactors as PDH and α-KGDH: TPP, Lipoate, CoA, FAD, NAD⁺. Leucine excess is most toxic (cerebral oedema, excitotoxicity). Presentation: neonatal encephalopathy, poor feeding, maple syrup odour. Treatment: dietary restriction of all 3 BCAAs + thiamine supplementation (some MSUD variants are thiamine-responsive).
8. Isoleucine (Ile, I)
Structure: Branched-chain aliphatic amino acid (BCAA); has 2 chiral centres
Key metabolic roles:
- One of the 3 BCAAs — catabolised in muscle
- Both glucogenic AND ketogenic:
- Glucogenic: produces Propionyl-CoA → (Biotin) Methylmalonyl-CoA → (B12) Succinyl-CoA (TCA entry)
- Ketogenic: produces Acetyl-CoA
- Important for haemoglobin structure: Ile E11 is a conserved residue in the haem pocket
- Catabolism blocked in MSUD (along with Leucine and Valine)
9. Valine (Val, V)
Structure: Branched-chain aliphatic amino acid (BCAA); smallest BCAA
Key metabolic roles:
- One of the 3 BCAAs — catabolised in muscle
- Purely glucogenic (of the 3 BCAAs) — produces Propionyl-CoA → Succinyl-CoA
- Critical structural role in haemoglobin: In sickle cell anaemia, the mutation is Glu6Val (glutamate at position 6 of β-globin is replaced by valine) — the hydrophobic valine causes HbS polymerisation under deoxygenated conditions
- Catabolism blocked in MSUD
10. Threonine (Thr, T)
Structure: Hydroxylated aliphatic amino acid; has 2 chiral centres; has a β-hydroxyl group
Key metabolic roles:
- Provides serine via threonine aldolase (minor pathway) or via dehydration
- Phosphorylation site on proteins (threonine kinases — major signalling) — serine/threonine kinases (e.g., PKA, PKC, mTOR) phosphorylate threonine residues for signal transduction
- Catabolism: multiple pathways producing pyruvate, succinyl-CoA, glycine — both glucogenic and ketogenic
- Abundant in intestinal mucin glycoproteins — structural role in mucosal barrier integrity
⚙️ Section 3 — Branched-Chain Amino Acids (BCAAs): A Special Focus
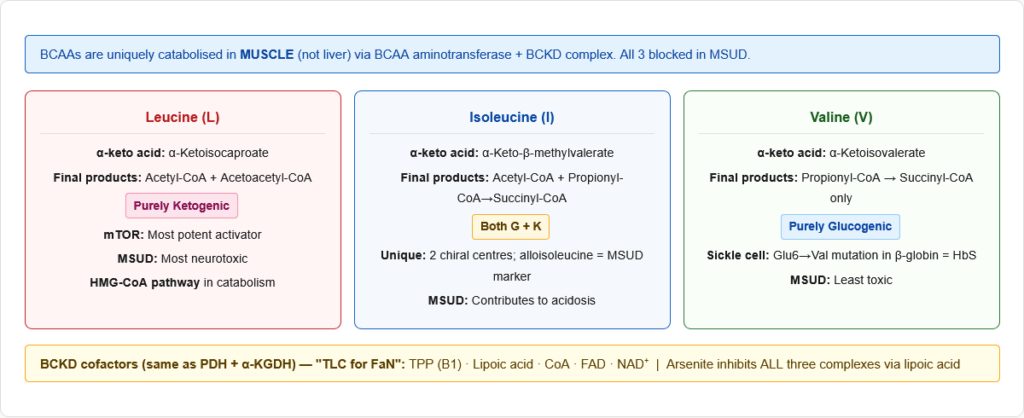
Why BCAAs Are Unique
The 3 BCAAs — Leucine, Isoleucine, Valine — share unique metabolic properties that distinguish them from all other amino acids:
1. Catabolism occurs primarily in MUSCLE (not liver) All other amino acids are mainly catabolised in the liver. BCAAs are catabolised in skeletal muscle, heart, kidney, and brain because:
- Muscle lacks the urea cycle (cannot dispose of nitrogen centrally)
- BCAAs serve as the primary nitrogen source for muscle glutamine synthesis
- The first enzyme (BCAA aminotransferase) is present in muscle but not significantly in liver
2. Nitrogen from BCAAs fuels the glucose-alanine cycle BCAA catabolism in muscle → releases NH₃ → transferred to pyruvate → alanine → exported to liver → urea cycle + gluconeogenesis
3. The BCKD complex is the rate-limiting step for all 3 BCAAs Branched-chain α-ketoacid dehydrogenase (BCKD) complex:
- Located in mitochondria
- Catalyses oxidative decarboxylation of all 3 BCAA-derived α-keto acids
- Same 5 cofactors as PDH and α-KGDH: TPP, Lipoic acid, CoA, FAD, NAD⁺
- Regulated by phosphorylation (BCKD kinase inhibits — analogous to PDH kinase)
- Deficiency: MSUD
BCAA catabolism summary:
| BCAA | α-Keto Acid | Final Products | Classification |
|---|---|---|---|
| Leucine | α-Ketoisocaproate | Acetyl-CoA + Acetoacetyl-CoA | Purely Ketogenic |
| Isoleucine | α-Keto-β-methylvalerate | Acetyl-CoA + Propionyl-CoA → Succinyl-CoA | Both |
| Valine | α-Ketoisovalerate | Propionyl-CoA → Succinyl-CoA | Purely Glucogenic |
🔬 Section 4 — One-Carbon Metabolism: The Folate-B12-Methionine Connection
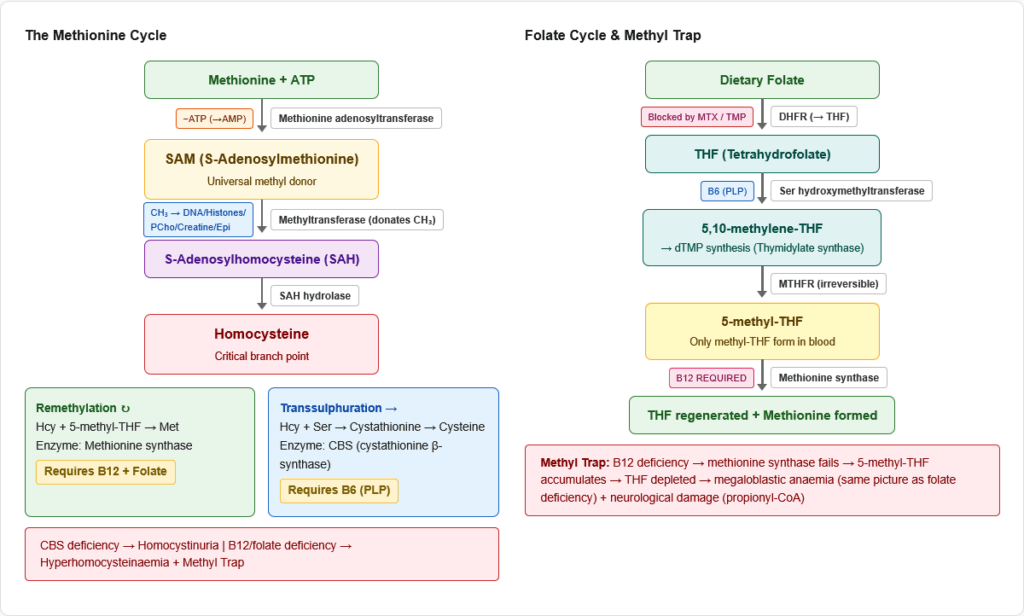
This is the most integrated and most-tested area of amino acid metabolism. It connects essential amino acids (methionine, histidine) to vitamin cofactors (B12, folate, B6) to clinical diseases (megaloblastic anaemia, neural tube defects, homocystinuria, MTHFR deficiency).
The Key Reactions
Folate cycle:
- Dietary folate → dihydrofolate (DHF) → tetrahydrofolate (THF) — enzyme: DHFR (target of methotrexate, trimethoprim)
- THF accepts 1-carbon units from serine: Serine + THF → Glycine + 5,10-methylene-THF — enzyme: Serine hydroxymethyltransferase (requires B6)
- 5,10-methylene-THF → 5-methyl-THF — enzyme: MTHFR (methylenetetrahydrofolate reductase) — irreversible
- 5-methyl-THF is the form that donates its methyl group to homocysteine to regenerate methionine — via methionine synthase (requires B12)
The methyl trap (B12 deficiency): When B12 is deficient, methionine synthase cannot function. 5-methyl-THF accumulates and cannot be converted back to THF (because the MTHFR reaction is irreversible). All folate becomes “trapped” as 5-methyl-THF → THF depletion → impaired DNA synthesis (same result as folate deficiency) → megaloblastic anaemia. This is why B12 deficiency causes megaloblastic anaemia even with normal folate intake.
Serine and Glycine — The One-Carbon Donors
- Serine is the primary donor of one-carbon units to the folate cycle
- Serine ↔ Glycine interconversion (serine hydroxymethyltransferase, B6) transfers a methylene group to THF
- Glycine + THF can also donate a carbon (glycine cleavage system — requires B6, lipoic acid, NAD⁺)
Summary of Cofactor Roles
| Vitamin/Cofactor | Reaction | Deficiency Consequence |
|---|---|---|
| Folate (THF) | 1-carbon carrier; DNA synthesis (dTMP) | Megaloblastic anaemia, NTDs |
| Vitamin B12 | Methionine synthase (homocysteine → methionine); Methylmalonyl-CoA mutase | Megaloblastic anaemia + neurological degeneration (subacute combined degeneration) |
| Vitamin B6 (PLP) | Transamination; serine hydroxymethyltransferase; CBS (Hcy → cystathionine); DOPA decarboxylase | Peripheral neuropathy; sideroblastic anaemia; homocystinuria (B6-responsive form) |
| Biotin | Carboxylation reactions (pyruvate carboxylase, ACC, propionyl-CoA carboxylase) | Multiple carboxylase deficiency |
🏥 Section 5 — Major Inborn Errors of Amino Acid Metabolism
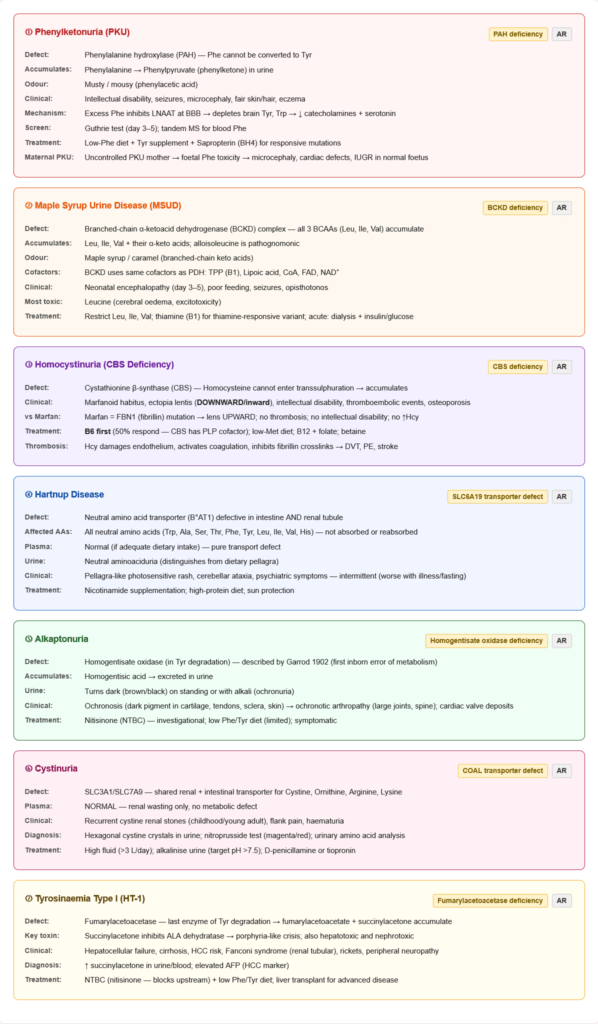
Phenylketonuria (PKU) — The Prototype
- Defect: Phenylalanine hydroxylase (PAH) deficiency (most common) or BH4 deficiency
- Inheritance: Autosomal recessive
- Biochemistry: Phe → cannot convert to Tyr → Phe accumulates → transaminated to phenylpyruvate → excreted (phenylketonuria)
- Why neurotoxic? Excess Phe competitively inhibits large neutral amino acid transporter (LNAAT) at BBB → reduces brain uptake of Tyr, Trp, Leu, Ile, Val → depletes catecholamines AND serotonin
- Clinical: Intellectual disability, seizures, microcephaly, fair complexion (reduced melanin), eczema, musty/mousy urine odour, behaviour problems
- Newborn screening: Guthrie test (bacterial inhibition assay — Phe inhibits bacterial growth reversal) or tandem mass spectrometry — done on day 3–5 of life
- Treatment: Low-phenylalanine diet (restrict Phe but not completely — it is essential), tyrosine supplementation, sapropterin (synthetic BH4) for BH4-responsive PAH mutations
- Maternal PKU: A phenylketonuric mother who is not on diet during pregnancy exposes the foetus to high Phe → foetal damage (microcephaly, cardiac defects, IUGR) — the foetus itself does not have PKU
Maple Syrup Urine Disease (MSUD)
- Defect: Branched-chain α-ketoacid dehydrogenase (BCKD) complex
- Inheritance: Autosomal recessive
- Biochemistry: All 3 BCAAs (Leu, Ile, Val) and their α-keto acids accumulate. Leucine excess → cerebral oedema, neuronal death (most toxic). Alloisoleucine (stereoisomer) is pathognomonic on plasma amino acid analysis.
- Clinical: Neonatal encephalopathy (lethargy, poor feeding, seizures), maple syrup/caramel urine odour from 3–5 days, opisthotonos, respiratory failure
- Newborn screening: Elevated Leu/Ile on tandem mass spectrometry
- Treatment: Dietary restriction of Leu, Ile, Val; leucine-free formula; thiamine supplementation (thiamine-responsive variant — E2 component mutation); acute episodes: haemodialysis, insulin + glucose (drives BCAAs into muscle)
Homocystinuria
- Defect: Cystathionine β-synthase (CBS) deficiency (most common), or B12/folate deficiency, or MTHFR deficiency
- Inheritance: Autosomal recessive (CBS deficiency)
- Biochemistry: Homocysteine accumulates → toxic to vascular endothelium, activates coagulation cascade, inhibits fibrillin cross-linking
- Clinical: Marfanoid habitus, ectopia lentis (downward/inward), intellectual disability, seizures, osteoporosis, thromboembolic events (DVT, PE, stroke — leading cause of death)
- Distinguishing from Marfan syndrome: Marfan = FBN1 mutation → upward/outward lens dislocation, no intellectual disability, no Hcy elevation. Homocystinuria = downward lens, intellectual disability, thrombosis, elevated homocysteine.
- Treatment: B6 supplementation (CBS has PLP cofactor; some mutations are B6-responsive → 50% of cases respond), low-methionine diet, B12 + folate (to push remethylation), betaine (alternative methyl donor)
Alkaptonuria
- Defect: Homogentisate oxidase (homogentisic acid oxidase) deficiency — in tyrosine degradation pathway
- Inheritance: Autosomal recessive (first inborn error of metabolism described by Archibald Garrod, 1902)
- Biochemistry: Homogentisic acid accumulates → excreted in urine → urine turns dark on standing/alkalinisation → deposits as dark pigment in collagen-rich tissues
- Clinical: Ochronosis (dark pigmentation of ear cartilage, sclera, intervertebral discs), arthritis (large joints, spine — ochronotic arthropathy), urine darkens on standing (ochronuria), cardiac valve deposits
- Diagnosis: Urine turns black with alkali or on air exposure; urinary homogentisic acid
- Treatment: Low tyrosine/phenylalanine diet (limited benefit); nitisinone (NTBC) being investigated; symptomatic management for arthritis
Hartnup Disease
- Defect: SLC6A19 (neutral amino acid transporter B°AT1) — defective intestinal and renal tubular transport of neutral amino acids (including Tryptophan, and large neutral amino acids)
- Inheritance: Autosomal recessive
- Biochemistry: Tryptophan cannot be absorbed from gut → tryptophan deficiency → niacin deficiency (since Trp is the precursor to niacin synthesis) → pellagra-like manifestations. Paradoxically, plasma Trp is low despite possible normal dietary intake because gut absorption fails.
- Clinical: Pellagra-like photosensitive rash, intermittent cerebellar ataxia, psychiatric symptoms, aminoaciduria (neutral amino acids in urine)
- Distinguishing from Pellagra: Hartnup = transport defect + aminoaciduria + normal dietary niacin. Pellagra = dietary niacin deficiency, no aminoaciduria.
- Treatment: Nicotinamide supplementation (bypasses the Trp→niacin pathway), high-protein diet, sun protection
Cystinuria
- Defect: Shared renal/intestinal transporter (SLC3A1/SLC7A9) for Cystine, Ornithine, Arginine, Lysine (COAL)
- Inheritance: Autosomal recessive
- Note: Plasma amino acid levels are NORMAL — this is purely a renal wasting disorder, not a metabolic defect
- Clinical: Recurrent cystine kidney stones (begin in childhood/early adulthood), renal colic, haematuria. Cystine is insoluble at normal urinary pH but soluble at pH >7.5.
- Diagnosis: Sodium nitroprusside test (positive — cystine turns red/purple); urinary amino acid analysis; hexagonal cystine crystals on urine microscopy
- Treatment: High fluid intake (>3 L/day), alkalinise urine with bicarbonate/acetazolamide (target pH >7.5), D-penicillamine (chelates cystine → more soluble mixed disulphide), tiopronin
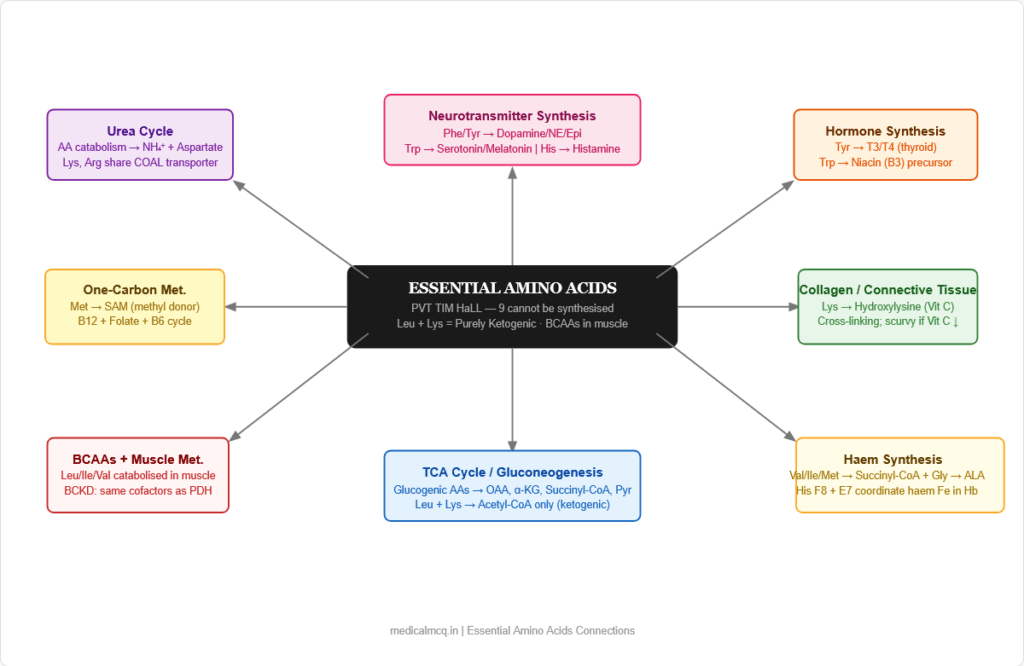
🔄 Section 6 — Connections to Other Pathways
→ Urea Cycle — Amino acid catabolism releases nitrogen as NH₄⁺ and aspartate, both of which enter the urea cycle. Histidine, glutamine, glutamate provide the bulk of nitrogen; aspartate (from OAA transamination) enters directly. Urea cycle disorders cause hyperammonaemia — the excess nitrogen cannot be disposed of.
→ TCA Cycle / Gluconeogenesis — All glucogenic amino acids produce TCA intermediates (OAA, α-KG, succinyl-CoA, fumarate) or pyruvate, feeding gluconeogenesis. During starvation, muscle protein catabolism provides the carbon skeletons for hepatic glucose synthesis via gluconeogenesis.
→ Neurotransmitter Synthesis — Phenylalanine/Tyrosine → catecholamines (dopamine, norepinephrine, epinephrine). Tryptophan → serotonin, melatonin. Histidine → histamine. Glutamate → GABA (glutamate decarboxylase, B6). Glycine → inhibitory neurotransmitter directly.
→ Haem Synthesis — Succinyl-CoA (from valine, methionine, isoleucine degradation via propionyl-CoA) + Glycine → δ-ALA (first step of haem synthesis, requires B6). Lysine provides hydroxylysine in haemoglobin crosslinks. Histidine coordinates the haem iron in haemoglobin (His F8, His E7).
→ One-Carbon Metabolism / DNA Synthesis — Methionine → SAM (methyl donor). Serine/glycine/histidine → THF 1-carbon units → dTMP synthesis (thymidylate synthase) → DNA replication. Folate and B12 deficiency impair this → megaloblastic anaemia.
→ Lipid Metabolism — Methionine (via SAM) methylates phosphatidylethanolamine → phosphatidylcholine (membrane phospholipid). Lysine (with methionine) → carnitine → fatty acid transport. Tryptophan → serotonin → regulates appetite/gut lipid absorption.
→ Collagen Synthesis — Lysine → hydroxylysine (lysyl hydroxylase, Vitamin C); Proline → hydroxyproline (prolyl hydroxylase, Vitamin C) → both essential for collagen triple helix stability and crosslinking. Vitamin C deficiency impairs both → scurvy.
🎯 High-Yield Exam Facts
These are the exact facts that appear repeatedly across NEET PG, USMLE, AIIMS and FMGE papers.
🔴 The 9 essential amino acids: PVT TIM HaLL (Phe, Val, Thr, Trp, Ile, Met, His, Leu, Lys) The single most-tested recall fact in amino acid biochemistry. Write this mnemonic until it is automatic.
🔴 Only 2 purely ketogenic amino acids: Leucine and Lysine All other amino acids are either purely glucogenic or both. “Leu and Lys — ketogenic ONLY.” These two CANNOT contribute to gluconeogenesis.
🔴 PKU = PAH deficiency → phenylalanine accumulates → phenylpyruvate in urine → musty odour Treatment: low-Phe diet + tyrosine supplementation. Sapropterin for BH4-responsive PKU. Maternal PKU damages foetus even though foetus itself doesn’t have PKU.
🔴 MSUD = BCKD deficiency → all 3 BCAAs accumulate → maple syrup odour → neonatal encephalopathy Same 5 cofactors as PDH: TPP, lipoic acid, CoA, FAD, NAD⁺. Leucine is most neurotoxic. Alloisoleucine is pathognomonic.
🔴 Homocystinuria = CBS deficiency → ectopia lentis DOWNWARD (vs Marfan = upward) + thromboembolism B6 treats 50% of cases. Distinguish from Marfan syndrome by: downward lens, intellectual disability, thrombosis, elevated homocysteine.
🟠 Vitamin B12 acts at methionine synthase (Hcy → Met) AND methylmalonyl-CoA mutase (methylmalonyl-CoA → succinyl-CoA) B12 deficiency causes: megaloblastic anaemia (methyl trap + impaired DNA synthesis) AND subacute combined degeneration of the cord (myelin instability from propionyl-CoA accumulation) AND elevated homocysteine AND elevated methylmalonic acid.
🟠 Alkaptonuria = homogentisate oxidase deficiency → urine darkens on standing → ochronosis → arthritis First inborn error of metabolism described by Garrod (1902). The urine darkening is the classic exam clue.
🟠 Hartnup disease = neutral amino acid transporter defect → pellagra-like rash + ataxia + aminoaciduria Treated with nicotinamide — bypasses the tryptophan → niacin pathway.
🟠 Cystinuria = COAL transporter defect → cystine stones → hexagonal crystals + nitroprusside test positive Plasma levels are NORMAL (renal wasting only). Target urine pH >7.5 (cystine soluble above this).
🟠 Tyrosinaemia Type I = fumarylacetoacetase deficiency → succinylacetone accumulates → inhibits ALA dehydratase → porphyria-like crises → HCC risk Treatment: NTBC (nitisinone) + dietary restriction. Succinylacetone is pathognomonic.
🟡 Vitamin C is required for BOTH lysyl hydroxylase AND prolyl hydroxylase → collagen cross-linking Scurvy = Vitamin C deficiency → defective collagen → bleeding gums, perifollicular haemorrhages, poor wound healing, corkscrew hairs.
🟡 Pellagra (niacin deficiency) can occur with isoniazid (INH) therapy — not just dietary deficiency INH inhibits pyridoxal phosphate-dependent enzymes, blocking tryptophan → niacin synthesis. Always supplement B6 with INH.
🟡 In sickle cell disease, the mutation is Glu→Val (position 6, β-globin) — a valine substitution creates hydrophobic patch for HbS polymerisation Glutamate (acidic, hydrophilic) replaced by Valine (nonpolar, hydrophobic) — a single essential amino acid substitution causes the entire disease.
🟡 The FIGLU excretion test diagnoses folate (and B12) deficiency via impaired histidine catabolism Histidine load → FIGLU cannot be converted (needs THF acceptor) → urinary FIGLU rises. High FIGLU = folate/B12 deficiency.
🧠 Mnemonics & Memory Tricks
“PVT TIM HaLL” — The 9 Essential Amino Acids → Helps you remember all 9 essential amino acids
P = Phenylalanine V = Valine T = Threonine T = Tryptophan I = Isoleucine M = Methionine H = Histidine L = Leucine L = Lysine
💡 Pro tip: PVT = Private (army rank); TIM = a name; HaLL = a room. Visualise a private named Tim standing in a hall. 9 letters = 9 amino acids.
“LOLA Likes Keto” — The Purely Ketogenic Amino Acids → Helps you remember that only Leucine and Lysine are purely ketogenic
L = Leucine O = Only two L = Lysine A = Are purely ketogenic
💡 Pro tip: Any exam question “which amino acid CANNOT contribute to gluconeogenesis?” = Leucine or Lysine. Every other amino acid has at least some glucogenic potential.
“COAL” — Cystinuria Transport Defect → Helps you remember which amino acids share the defective transporter in cystinuria
C = Cystine O = Ornithine A = Arginine L = Lysine
💡 Pro tip: Cystine is the insoluble one that forms stones — that’s the clinical problem. The others are lost in urine but don’t stone because they’re soluble. The question always shows hexagonal crystals or a nitroprusside test result.
“TLC for FaN” — Cofactors of BCKD (Same as PDH and α-KGDH) → Applies to all three multi-enzyme dehydrogenase complexes (PDH, α-KGDH, BCKD)
T = TPP (Thiamine B1) L = Lipoic acid C = CoA F = FAD N = NAD⁺
💡 Pro tip: Any question involving MSUD, thiamine-responsive variant, or “same cofactors as pyruvate dehydrogenase” is testing this shared cofactor set. Arsenite inhibits ALL three complexes (via lipoic acid binding).
Amino Acid → Neurotransmitter Quick Map: Phe/Tyr → Catecholamines (Dopamine, NE, Epi) | Trp → Serotonin, Melatonin | His → Histamine | Glu → GABA (B6) | Gly → Glycine (inhibitory NT)
💡 Pro tip: “What is the precursor to serotonin?” = Tryptophan. “What is the precursor to dopamine?” = Tyrosine (from Phenylalanine). Rate-limiting enzyme for both = a hydroxylase requiring BH4.
⚠️ Common Mistakes Students Make on This Topic
❌ Mistake: “Tyrosine is an essential amino acid” ✅ Reality: Tyrosine is NON-essential under normal conditions because it is synthesised from phenylalanine by PAH. It becomes conditionally essential only in PKU (when PAH is deficient). In PKU patients, tyrosine must be supplemented in the diet — making it essential for them. 📝 How this gets tested: “Which amino acid becomes essential in PKU?” — answer is Tyrosine, not because it is normally essential, but because the synthetic pathway from phenylalanine is blocked.
❌ Mistake: “Leucine can contribute to gluconeogenesis because it produces Acetyl-CoA” ✅ Reality: Acetyl-CoA CANNOT contribute to net gluconeogenesis in mammals — its carbons are lost as CO₂ in the TCA cycle. Leucine is the most purely ketogenic amino acid and cannot contribute even partially to glucose synthesis. Contrast with Isoleucine, which produces BOTH Acetyl-CoA (ketogenic) AND propionyl-CoA → succinyl-CoA (glucogenic). 📝 How this gets tested: “A patient is in prolonged starvation — which amino acid contributes most to gluconeogenesis?” — Leucine is the WRONG answer. Alanine, glutamine, and aspartate are the main contributors. Leucine can only make ketone bodies.
❌ Mistake: “In homocystinuria and Marfan syndrome, the lens dislocation is in the same direction” ✅ Reality: In Marfan syndrome (FBN1 mutation — fibrillin defect), the lens dislocates upward and outward (superotemporal). In homocystinuria (CBS deficiency), the lens dislocates downward and inward (inferonasal). The direction is the classic exam differentiator. 📝 How this gets tested: A clinical scenario describes a tall patient with long limbs and dislocated lens — the direction of dislocation + presence of intellectual disability + thrombosis will distinguish homocystinuria from Marfan syndrome.
❌ Mistake: “Hartnup disease is simply pellagra” ✅ Reality: Hartnup disease is a transporter defect causing tryptophan malabsorption, which secondarily causes niacin deficiency (pellagra-like symptoms). But Hartnup disease also causes aminoaciduria (neutral amino acids in urine), cerebellar ataxia, and responds to nicotinamide rather than niacin. Crucially, dietary niacin intake may be normal — the problem is Trp absorption. 📝 How this gets tested: “A patient with photosensitive rash, ataxia, and neutral amino acids in urine but normal plasma levels” — this is Hartnup, not pellagra. The aminoaciduria distinguishes them.
❌ Mistake: “B12 and folate deficiency both cause only megaloblastic anaemia” ✅ Reality: B12 deficiency causes megaloblastic anaemia PLUS neurological damage (subacute combined degeneration of the spinal cord — demyelination of dorsal columns and lateral corticospinal tracts) because B12 is also needed for methylmalonyl-CoA mutase — propionyl-CoA cannot be metabolised → abnormal fatty acids incorporated into myelin → neurological damage. Folate deficiency causes megaloblastic anaemia only — NO neurological disease. 📝 How this gets tested: “Megaloblastic anaemia + peripheral neuropathy + posterior column signs” = B12 deficiency (not folate). Treating B12 deficiency with folate alone will correct the blood picture but WORSEN the neurological disease — this is the classic clinical trap.
📝 5 Practice MCQs — Test Yourself Now
Q1: A 6-week-old infant is referred after a positive newborn screen. Plasma phenylalanine is 18 mg/dL (normal <2). The child appears normal. The mother asks why tyrosine must be added to the special formula. What is the correct explanation?
- A. Tyrosine is an essential amino acid required for all protein synthesis
- B. The restricted phenylalanine diet means the child cannot synthesise tyrosine from phenylalanine, making it conditionally essential
- C. Tyrosine competes with phenylalanine for intestinal absorption and must be given to correct the ratio
- D. Tyrosine directly inhibits phenylalanine hydroxylase, preventing further phenylalanine accumulation
✅ Answer: B. The restricted phenylalanine diet means the child cannot synthesise tyrosine from phenylalanine, making it conditionally essential
Why correct: Tyrosine is normally synthesised from phenylalanine by phenylalanine hydroxylase (PAH). In PKU, PAH is deficient, so this pathway is blocked. In addition, the PKU diet severely restricts phenylalanine intake (eliminating the precursor), so no tyrosine can be synthesised. Without supplementation, tyrosine deficiency would impair catecholamine synthesis, thyroid hormone production, and melanin synthesis — worsening the clinical picture beyond just phenylalanine accumulation.
Why A is wrong: Tyrosine is NOT a standard essential amino acid — it is non-essential under normal conditions precisely because it can be made from phenylalanine. Calling it essential for all protein synthesis is incorrect. Why C is wrong: While amino acids do share transporters, tyrosine supplementation in PKU is specifically to replace the deficit caused by both blocked synthesis and dietary restriction — not to adjust absorption competition. Why D is wrong: Tyrosine does not inhibit PAH. In fact, in classic PKU, PAH has no functional activity regardless of tyrosine levels.
Exam tip: “Which amino acid becomes essential in PKU?” = Tyrosine. This question directly tests that concept. PKU also means melanin is reduced (light skin/hair), catecholamines are reduced (because Tyr is the precursor), and thyroid hormone synthesis may be impaired.
Q2: A 4-day-old neonate in the NICU has poor feeding, lethargy, and seizures. The urine has a distinctive sweet, caramel odour. Plasma amino acid analysis shows markedly elevated leucine, isoleucine, and valine, with a peak corresponding to alloisoleucine. Which cofactor set does the deficient enzyme share with pyruvate dehydrogenase?
- A. Biotin, Lipoic acid, CoA, FAD, NAD⁺
- B. TPP, Lipoic acid, CoA, FAD, NAD⁺
- C. TPP, Biotin, CoA, FAD, NADP⁺
- D. PLP, Lipoic acid, CoA, FMN, NAD⁺
✅ Answer: B. TPP, Lipoic acid, CoA, FAD, NAD⁺
Why correct: This is MSUD (Maple Syrup Urine Disease) caused by BCKD (branched-chain α-ketoacid dehydrogenase) deficiency. The BCKD complex shares the exact same 5 cofactors as PDH and α-KGDH: TPP (Thiamine pyrophosphate), Lipoic acid, CoA, FAD, and NAD⁺. This mnemonic — TLC for FaN — applies to all three multi-enzyme oxidative decarboxylase complexes. This is why thiamine-responsive MSUD variants exist.
Why A is wrong: Biotin replaces TPP in this option. Biotin is the cofactor for carboxylation reactions (pyruvate carboxylase, ACC) — NOT for oxidative decarboxylation complexes. TPP is the correct cofactor for decarboxylation in PDH, α-KGDH, and BCKD. Why C is wrong: Biotin and NADP⁺ are both wrong. BCKD uses NAD⁺ (producing NADH), not NADP⁺. And biotin is not in this complex. Why D is wrong: PLP (pyridoxal phosphate/B6) is the cofactor for transamination and amino acid decarboxylation, not for the multi-enzyme oxidative decarboxylase complexes. FMN is in Complex I of ETC, not in these dehydrogenase complexes.
Exam tip: The shared cofactor set “TLC for FaN” is tested by asking about MSUD, PDH, or α-KGDH in three different disease contexts. Arsenite poisons all three by binding lipoic acid (the L in TLC).
Q3: A 22-year-old woman presents with recurrent deep vein thromboses and is found to have a dislocated lens on slit-lamp examination. She has mild intellectual disability and a tall, thin frame. Plasma homocysteine is 180 µmol/L (normal <15). Genetic testing shows a CBS mutation. Administration of which vitamin would be the most appropriate first-line treatment to try?
- A. Vitamin B12 (cobalamin)
- B. Vitamin B6 (pyridoxine)
- C. Vitamin B9 (folic acid)
- D. Vitamin B1 (thiamine)
✅ Answer: B. Vitamin B6 (pyridoxine)
Why correct: This is classical homocystinuria from cystathionine β-synthase (CBS) deficiency. CBS is a pyridoxal-5-phosphate (PLP/B6)-dependent enzyme. Approximately 50% of CBS-deficient patients have a B6-responsive mutation — their CBS has a reduced affinity for PLP, and pharmacological doses of B6 can restore sufficient activity to normalise homocysteine. B6 supplementation is always tried first in CBS deficiency because it is safe, cheap, and works in half the cases.
Why A is wrong: B12 is needed for methionine synthase (remethylation pathway). It lowers homocysteine in B12 deficiency or in the remethylation defects (MTHFR deficiency), but CBS deficiency is a transsulphuration defect — B12 alone is insufficient and not first-line for CBS mutations. Why C is wrong: Folate (B9) drives the remethylation of homocysteine to methionine via MTHFR and methionine synthase. It can help lower homocysteine in combination therapy but is not the primary treatment for CBS deficiency. Why D is wrong: Thiamine (B1) is the cofactor for pyruvate dehydrogenase, α-KGDH, and BCKD — none of which involve homocysteine metabolism. B1 is relevant in MSUD and PDH deficiency, not homocystinuria.
Exam tip: CBS deficiency → B6 trial first (50% respond). If non-responsive → low-methionine diet + B12 + folate + betaine. The combination treats both the transsulphuration failure (diet) and remethylation pathway enhancement (B12/folate/betaine).
Q4: A 25-year-old man with a history of recurrent renal calculi presents with right-sided flank pain and haematuria. Urinalysis shows hexagonal crystals. Urine sodium nitroprusside test is positive. Plasma amino acid levels are normal. What is the underlying defect?
- A. Overproduction of cystine from methionine due to enzyme hyperactivity
- B. Defective renal tubular and intestinal transporter for cystine, ornithine, arginine, and lysine
- C. Deficiency of cystathionine β-synthase preventing cysteine catabolism
- D. Increased dietary cystine absorption due to upregulated intestinal cystine transporter
✅ Answer: B. Defective renal tubular and intestinal transporter for cystine, ornithine, arginine, and lysine
Why correct: This is cystinuria — caused by mutations in the SLC3A1/SLC7A9 genes encoding the shared amino acid transporter for Cystine, Ornithine, Arginine, and Lysine (COAL). The transporter failure causes urinary loss of all four amino acids (aminoaciduria) but PLASMA levels are normal because hepatic synthesis and metabolism are unaffected. Cystine is poorly soluble in urine (particularly at normal urinary pH) and precipitates as hexagonal crystals → renal stones. The sodium nitroprusside test turns magenta/red with cystine.
Why A is wrong: There is no enzymatic overproduction of cystine in this condition. The problem is entirely one of tubular reabsorption failure, not metabolic overproduction. Cystine production from methionine via CBS is normal. Why C is wrong: CBS deficiency causes homocystinuria (elevated homocysteine, intellectual disability, lens dislocation, thrombosis) — not cystine stones. CBS deficiency would impair cysteine SYNTHESIS from homocysteine, which would lower cystine — the opposite of cystinuria. Why D is wrong: In cystinuria, the intestinal transporter is DEFECTIVE (not upregulated), meaning cystine is UNDER-absorbed from the gut — not over-absorbed. The aminoaciduria is from renal tubular failure.
Exam tip: “Hexagonal crystals + nitroprusside positive + recurrent stones in young patient + normal plasma amino acids” = Cystinuria every time. Distinguish from homocystinuria (plasma Hcy elevated, no crystals, no stones).
Q5: A 19-year-old student presents episodically with a pellagra-like rash on sun-exposed areas, ataxia, and psychiatric symptoms. She reports that the episodes improve when she is at home eating well but worsen during exam stress. Urine amino acid electrophoresis shows increased neutral amino acids. Plasma levels are normal. Niacin intake is adequate. What is the most likely diagnosis and mechanism?
- A. Pellagra from dietary niacin deficiency worsened by physiological stress
- B. Hartnup disease — defective intestinal and renal neutral amino acid transporter → tryptophan deficiency → reduced niacin synthesis
- C. Carcinoid syndrome — excess serotonin production from tryptophan depletes niacin
- D. MTHFR deficiency — impaired one-carbon metabolism affecting niacin regeneration
✅ Answer: B. Hartnup disease — defective intestinal and renal neutral amino acid transporter → tryptophan deficiency → reduced niacin synthesis
Why correct: This is classic Hartnup disease. The defective SLC6A19 transporter (B°AT1) impairs intestinal absorption AND renal reabsorption of neutral amino acids (including tryptophan). With tryptophan deficiency, the body cannot synthesise adequate niacin from Trp (normally 60 mg Trp → 1 mg niacin), causing pellagra-like symptoms. The intermittent nature (worse during physiological stress/illness when amino acid demands increase) and the urine showing neutral aminoaciduria with normal PLASMA levels are pathognomonic. Dietary niacin being adequate rules out simple pellagra.
Why A is wrong: Simple pellagra from dietary deficiency would not cause urinary neutral amino acid spillage, and the niacin intake is stated to be adequate. Stress worsens Hartnup (increased AA demands) but wouldn’t cause aminoaciduria in dietary pellagra. Why C is wrong: Carcinoid syndrome causes excess serotonin synthesis FROM tryptophan (diverting Trp away from niacin), causing pellagra-like features. However, carcinoid presents with episodic flushing, diarrhoea, bronchospasm, and right-sided heart valve lesions — not ataxia or aminoaciduria. Urinary 5-HIAA would be elevated. Why D is wrong: MTHFR deficiency impairs folate one-carbon metabolism and causes hyperhomocysteinaemia. It has no direct role in tryptophan/niacin metabolism and does not cause neutral aminoaciduria or pellagra-like symptoms.
Exam tip: Hartnup disease diagnostic triad: (1) Pellagra-like rash + ataxia (2) Neutral aminoaciduria with NORMAL plasma levels (3) Responds to nicotinamide supplementation. If any two of these are in the vignette, Hartnup should be your first thought.
📚 References
📖 Harper’s Illustrated Biochemistry — Rodwell et al. | Chapter 26: Catabolism of Proteins & of Amino Acid Nitrogen; Chapter 27–29: Catabolism of the Carbon Skeletons of Amino Acids
📖 Lippincott’s Illustrated Reviews: Biochemistry — Ferrier | Chapter 20: Amino Acid Degradation and Synthesis; Chapter 21: Conversion of Amino Acids to Specialised Products
📖 Lehninger Principles of Biochemistry — Nelson & Cox | Chapter 18: Amino Acid Oxidation and the Production of Urea
📖 Stryer’s Biochemistry — Berg, Tymoczko & Stryer | Chapter 23: Protein Turnover and Amino Acid Catabolism
📖 Vasudevan & Sreekumari’s Textbook of Biochemistry — Vasudevan | Chapter 13: Metabolism of Amino Acids
📖 Robins Basic Pathology — Kumar et al. | Chapter 5: Genetic Disorders (Inborn errors of amino acid metabolism)
🚀 Keep Practising — You Are Not Done Yet
Amino acid metabolism questions in NEET PG and USMLE nearly always come as clinical vignettes — a child with odd-smelling urine, a young adult with lens dislocation, a patient with recurrent kidney stones, a neonate with lactic acidosis. The biochemistry is the decoder ring for the clinical picture.
Recognising the pattern — odour + neurological = MSUD; musty odour + fair skin = PKU; downward lens + thrombosis = homocystinuria; hexagonal crystals + young patient = cystinuria — is what clinical biochemistry is all about.
medicalmcq.in has free Biochemistry MCQs covering every inborn error of amino acid metabolism in this article, with full clinical-scenario format and detailed mechanistic explanations.
Work through the amino acid metabolism set immediately after reading this article. Pattern recognition comes from volume of practice — not from re-reading.